- It was long assumed, wrongly as it happens, that gene expression was regulated exclusively by activators and repressors of transcription operating on noncoding regulatory elements within a given gene.
- More recently, it has been noted that gene expression is subject to multiple additional levels of control, all of which may become – in some way – defective in cancer. Thus, in addition to mutations in coding DNA and well-known regulatory elements, there are seemingly endless opportunities for an evolving cancer cell to skew gene and protein expression to serve its own ends, including:
- Chromatin modifications that are transmitted from one somatic cell to all its descendants. Such a control is referred to as “epigenetic,” as the DNA sequence is not altered.
- Posttranscriptional modification of mRNA.
- Stability and processing of mRNA.
- Regulation of genes and mRNAs by small RNA molecules encoded by areas of the genome previously believed to be noncoding “junk.”
- Chromatin modifications that are transmitted from one somatic cell to all its descendants. Such a control is referred to as “epigenetic,” as the DNA sequence is not altered.
- Patterns of DNA methylation and chromatin structure are often markedly disturbed in cancer cells potentially resulting in inappropriate silencing of tumor suppressor genes (hypermethylation) or conversely activation of oncogenes (hypomethylation). This can arise through the action of mutations in epigenetic regulators and environmental factors.
- Some cancers may be epigenetically unstable. Such cancers exhibit hypermethylation of usually unmethylated regions such as the CpG islands, found in up to half of all human gene promoters (CpG island methylator phenotype (CIMP)), resulting in aberrant silencing of hundreds of genes. CIMP is analogous to the mutator phenotype resulting from defects in DNA repair described in Chapter 10.
- It is now generally accepted that epigenetic modification can be inherited by the progeny of somatic cells, and therefore silencing of genes by this means will be passed on through successive generations of evolving cancer cells alongside any coincident alterations in the coding sequence of the DNA.
- It was also taken for granted that the epigenetic “slate” would be wiped clean in germ cells and at fertilization so that these experience-driven traits would not be passed on to successive generations of the organism.
- This view has, however, had to adapt in order to account for several observations which are hard to explain by genetic inheritance alone:
- Firstly, the relationship between inadequate maternal nutrition and unhealthy lifestyle on the subsequent risk of heart disease and diabetes in the offspring. Although long ignored by the mainstream, one might say “the elephant in the womb,” it is now generally accepted that fetal conditioning in utero can permanently alter metabolism and disease expectation for the child later in life.
- Secondly, exciting recent studies suggesting that a father’s experiences might exert a lasting influence on disease risk in the offspring are very hard to explain other than by the transfer of epigenetic information.
- Firstly, the relationship between inadequate maternal nutrition and unhealthy lifestyle on the subsequent risk of heart disease and diabetes in the offspring. Although long ignored by the mainstream, one might say “the elephant in the womb,” it is now generally accepted that fetal conditioning in utero can permanently alter metabolism and disease expectation for the child later in life.
- Several miRNAs are aberrantly expressed in human cancers, where they can act as tumor suppressors or sometimes oncogenes and constitute an important new class of treatment targets and biomarkers. Endogenous small interfering RNAs (siRNAs) are broadly more homologous to specific mRNAs than their miRNA counterparts. As a result, siRNAs regulate individual genes whereas miRNAs may operate as master switches controlling multiple genes in a coordinated fashion. The endogenous siRNAs are comparatively less well studied, but this mechanism is being exploited in the design of a range of novel therapies.
- Proteins are subject to a range of posttranslational modifications, including phosphorylation, ubiquitination, and sumoylation, which strongly influence their activity, stability, and location and may become aberrant in cancer cells.
- The ubiquitin–proteasome pathway is involved in degradation of numerous cancer-relevant intracellular proteins, including regulators of apoptosis and cell cycling. Mutations in ligases and other genes may compromise protein stability and degradation and contribute to tumorigenesis. Inhibitors of the proteasomal degradation of proteins are being examined as therapeutic agents against cancer.
- Aside from degradation, the biological activity of proteins may also be influenced by localization and partitioning within the cell.
Introduction
The discovery of oncogenes in the 1970s provided strong support for the contention that cancer was a genetic disease. Gradually this extended into a pervasive notion that within every cancer cell, and written into its genome, was the autobiography that would explain to anyone with the tools to read it exactly how a normal cell, against all the odds, achieved immortality. Every step in its evolution would be faithfully recorded from the precious first edition through all subsequent editions. Thus, even if the older editions become rare or out of print, their legacy remains visible within the most current. Thus, key events in the evolution of the cancer cell could be identified, and treatments and preventative strategies developed based on this knowledge. However, this requires the necessary tools – imagine having your ebook but no suitable reader. The human genome project and advances in systems biology have already allowed many of these cancer biographies to be read, and although much useful information has been acquired it has become very clear that the sequence of the DNA text is only part of the story. To avoid torturing this metaphor any further, we will conclude by noting that to fully comprehend what the biography means, we must also have the epigenetic “punctuation.”
Every living being is also a fossil. Within it, all the way down to the microscopic structure of its proteins, it bears the traces if not the stigmata of its ancestry.
Jacques Monod
This chapter will explain how cancers may arise by changes in the expression of genes and proteins that are not primarily the result of alterations in the DNA sequence of protein-coding genes. Because mutations cause changes in expression of proteins, it does not follow that increased or reduced expression of a given gene or protein can result only from alteration to the DNA sequence. Rather, as we will see, chemical alteration of the DNA or of important associated proteins also has potent effects on gene expression, without any alteration of the DNA code. On the other hand, the genes that encode the regulators of methylation, acetylation, and other key processes that can influence gene expression are also subject to mutation. These mutations have the potential to influence expression of large numbers of genes by altering methylation patterns or expression of regulatory factors involved in protein translation and stability. Recent important examples of epigenetic changes resulting from mutations in regulatory genes include those in ARID1A and in numerous other genes encoding a variety of DNA methyltransferases, members of the SWI–SNF and miRNAs. However, in these cases the mutation is not in the gene encoding the protein concerned. This is not an abstract or semantic issue as exemplified by the long-overlooked role of MYC protein deregulation in the majority of cancers where, in complete contrast to Burkitt lymphoma, no chromosomal rearrangements or mutations in the MYC gene are present.
With the completion of genome-sequencing projects, one of the major challenges in modern biology is to understand what all the tens of thousands of identified genes actually do and moreover how their activity is regulated. To face this considerable challenge will require a much greater understanding of all the many processes that contribute to the regulation of gene expression and the ultimate formation of functional protein products. It has long been appreciated that gene expression is regulated by protein complexes that either promote or repress expression by binding to specific regulatory elements adjacent to but distinct from the coding region. We have also extensively discussed how gene expression is altered by mutations in these or the coding regions. One of the most exciting areas of cancer research now is describing and trying to interfere with other mechanisms, many alluded to in earlier chapters, which together conspire to influence whether a protein is made or not and how amounts in the cell are regulated. In fact, generation of functional proteins is determined by a remarkable variety of processes, many only recently appreciated. The term epigenetics (meaning “in addition to” genetics) was first used in the 1940s by Conrad Waddington to encompass nongenetic influences on phenotype. Epigenetics is now specifically used to describe stable mitotic or even meiotic inheritance of phenotype resulting from changes in a chromosome without alterations in the DNA sequence – in other words, non-Mendelian but heritable modifications such as chromatin remodeling that can alter gene expression in a cell and in its progeny.
Other important regulatory processes have come to light over the last decade. Once the gene has been transcribed, but before the mRNA is translated into protein synthesis, the gene transcripts are subject to alternative splicing, the inhibitory effect of noncoding RNAs (ncRNAs), and other factors affecting stability. Finally, translation of mRNA into protein is regulated at the ribosomes, and the proteins themselves are further modified in a bewildering number of ways that affect protein function, localization, and longevity. In fact, as the final functional arbiters, the proteins are subject to a variety of regulatory processes and when their usefulness has expired can be summarily destroyed or alternatively exiled, segregated, or excluded in order to prevent any further interactions with other proteins.
Without belaboring the point, as this has been covered in depth in this book, tumor cells are characterized by aberrant responses to cellular signals that normally regulate cell replication, differentiation, adhesion and motility, and apoptosis. As discussed in other chapters, these cellular processes are regulated by proteins and thereby by all of the aforementioned regulatory processes. In fact, essentially all links in the chain leading from an accurately maintained genome, through gene transcription and translation and ultimately protein modification, are in some way subject to gene mutations, and potentially cancer-causing mutations at that. But changes in gene and protein expression do not only arise as a result of gene mutations. In fact, protein levels can also be regulated by numerous other relatively more recently described processes operating at different stages of the gene–protein sequence, including:
- Changes in the methylation and structure of the DNA;
- Acetylation and numerous other posttranslational modifications of the associated histone proteins, which remodel chromatin;
- Alternative splicing of mRNA;
- Noncoding RNAs such as miRNAs; and
- Stability, transport, and partitioning of the key functional effectors – the proteins themselves.
Methylation of cytosine is a critical epigenetic alteration with profound regulatory effects on both the transcription and the replication of DNA and is also very well preserved through cell divisions. Methylation of a gene is effectively acting as a “mute button” that silences expression of that gene. Patterns of DNA methylation, which are passed on to cellular progeny, are responsible for achieving cellular differentiation and tissue-specific gene expression amongst cells which broadly all have identical genomes. Appreciation of the pivotal role played by methylation in cell biology and in particular how this can lead to imprinting of genes as well as the silencing of tumor suppressor genes during tumorigenesis have fueled a major research initiative into finding drugs that target this process. The main enzymes regulating methylation are the DNA methyltransferases (DNMTs), which can add methyl groups to the 5′ position of cytosine rings within CpG dinucleotides. The other key epigenetic modification with powerful effects on control of gene expression is acetylation of histones. The dynamics of histone acetylation is dictated by balancing the opposing actions of histone acetyl transferases (HATs) and histone deacetylases (HDACs). In general, HATs activate and HDACs silence gene expression – “Stand still and silent with your HAT off and your HDAC on.”
HDACs are important epigenetic regulators of gene expression through the remodeling of chromatin and have been discussed in other chapters in the context of c-MYC (Chapter 6) and RB (Chapter 4). It is important to be aware that histones are susceptible to the whole panoply of posttranslational modifications, and acetylation is just one of these. Methylation, phosphorylation, ubiquitination, and sumoylation are all present and may all contribute to chromatin structuring in one way or another. By implication, therefore, the enzymes that mediate the addition or removal of these modifications play important roles in normal cell processes and in cancer, and could all be potential drug targets. The combination of all of these various modifications in a given genomic region and the resultant effects on chromatin confirmation and gene expression have been dubbed the histone code. Another term that has become commonplace is epigenome, which refers to the sum of all epigenetic factors operating within a given cell at a particular time.
Histone modifications and methylation of cytosine are vulnerable to the effects of differing exogenous agents, including certain base analogues, radiation, tobacco smoke, hormones, and reactive oxygen species (see also Chapter 3), all of which can potentially influence gene expression and cellular phenotype epigenetically (methylation and/or acetylation, particularly of CpG islands in gene promoter regions) without changing their DNA sequence. It is worth noting that most of these factors could also cause mutations. Cancer cells typically manifest profound alterations in DNA methylation and histone modification patterns, including global hypomethylation and promoter-specific hypermethylation of DNA. Intriguingly, such epigenetic changes may already, in the main, be established in pre-malignant stages, powerfully arguing a case for causality in many human cancers. The mechanisms are becoming clear. Firstly, global hypomethylation can precipitate genomic instability by accelerating chromosomal rearrangements and translocations and could moreover activate large numbers of oncogenes and release imprinting of growth factors, such as IGF-2. Secondly, CpG island promoter hypermethylation could, as already discussed in Chapter 7, provide a “second hit” by silencing the remaining functioning allele of a tumor suppressor gene or miRNA. Don’t worry – mutations are not off the hook here; the methylase-encoding gene DNMT3A is mutated in some cancers such as AML, and can contribute to the establishment of an aberrant cancer epigenome.
Over the last decade, both academic and industrial sectors have been driven to a frenzy of research activity, takeovers, and buyouts, prompted by the discovery that what was once embarrassingly referred to as “junk DNA” actually contains the blueprint for generating a new family of key regulatory RNA molecules. After a hasty rebranding exercise, what we now refer to as “noncoding DNA” (DNA that is not ultimately translated into protein) has been shown to encode an ever-expanding family of important regulatory factors, the imaginatively named noncoding RNAs (ncRNAs) that include the all-conquering microRNAs (miRNAs). So, far from being worthless junk, much of the genome is actually transcribed into thousands of ncRNAs, including not only miRNAs but also small interfering RNAs (siRNAs) and a variety of long ncRNAs that impose powerful transcriptional and posttranscriptional controls on protein synthesis. In fact, to show just how wide of the mark we were, miRNAs alone regulate at least 30% of all human genes by controlling translation and degradation of target mRNAs. Interestingly, the DNA encoding miRNAs is also subject to regulation by methylation – a form of “epi-epigenetics.”
Aside from regulation of gene expression, the levels of oncoproteins and tumor suppressor proteins may also become abnormal through either increased or decreased degradation (Chapters 6 and 7). Lysosomes were historically regarded as the principal means of protein degradation within the cytoplasm. However, over the last 10–15 years, attention has increasingly focused on the role of ubiquitination of intracellular proteins; such ubiquitinated proteins are thereby targeted for degradation by a multiprotein complex termed the proteasome. In general it now seems that extracellular and transmembrane proteins are primarily degraded in the lysosomes, whereas intracellular proteins, including key regulators of the cell cycle and apoptosis, are normally degraded by the proteasome. Self-evidently, many proteasome substrates are involved in pathways that become deregulated in cancer, and proteasome inhibitors are now entering clinical practice. The partitioning and localization of proteins are also important determinants of biological activity, and these will also be discussed in this chapter.
As we have done in other chapters, we will start with a brief refresher on relevant cell biology which will make the mechanics of epigenetic regulation much easier to follow.
The Language of Epigenetics
Everything in life is speaking in spite of its apparent silence.
Hazrat Inayat Khan
Chromatin
DNA does not exist naked within the cell, but in association with histones, which constitute a protein scaffold that gives form to the complex tertiary structure referred to as chromatin (see Box 11.1). Chromatin was originally observed to exist in two different forms, during microscopic examination of cells during interphase. These observational differences are now known to correlate with gene expression activity – heterochromatin represents repressed segments of more tightly packed chromosomal DNA, while euchromatin represents a more open configuration with transcriptionally active segments. These two forms of DNA are interconvertible. During cell differentiation and maturation, RNA synthesis declines and is accompanied by a corresponding conversion of euchromatin to heterochromatin – therefore, fewer genes are available for mRNA synthesis. A wide variety of cellular processes involve de-repression of previously repressed genes, and cells undergoing such gene de-repression often display a reversible transformation of heterochromatin to euchromatin within their nuclei. A similar alteration in DNA takes place during mitosis. At the onset of prophase, the nuclear membrane dissolves and the euchromatin seen during interphase condenses into large chromosomal masses in prelude to metaphase when the chromosomes will eventually become segregated and separated for completion of cell division. Strictly (although very similar at a molecular-level reduced mRNA synthesis), this condensation of interphase euchromatin into condensed chromosomal masses is not classed as heterochromatin because this terminology was restricted to describing appearances of chromosomes during interphase.
Nucleosomes form the basic units of chromatin, and each comprises 146 base pairs of DNA wrapped around an octamer of two molecules each of the histones H2A, H2B, H3, and H4. Neighboring nucleosomes are joined to each other by linker DNA, and progressive coiling of nucleomes leads to higher order structures. DNA contained within compacted chromatin, known as heterochromatin, is not available for transcription, unless appropriate activation of remodeling processes first takes place in order to enable access of the transcription factors and transcriptional machinery to individual genes. Chromatin remodeling requires the action of two classes of proteins: those that covalently modify DNA or histones (by methylation, acetylation, etc.) and those that mobilize nucleosomes such as the SW1–SNF complex.
SWI–SNF chromatin-remodeling complexes require energy generated by hydrolysis of ATP in order to influence gene expression through nucleosome remodeling. As will be discussed, these complexes function as tumor suppressors and are frequently inactivated in human cancers (see Table 11.1 for a list of SWI component-encoding genes altered in cancers). These include a variety of tumors suppressors such as BRG1, BRM, ARID1A, SMARCC1, and SMARCB1 that are often inactivated in human cancers.
Table 11.1 List of genes encoding epigenetic regulators, known to be compromised by deregulated expression or mutation in human cancers
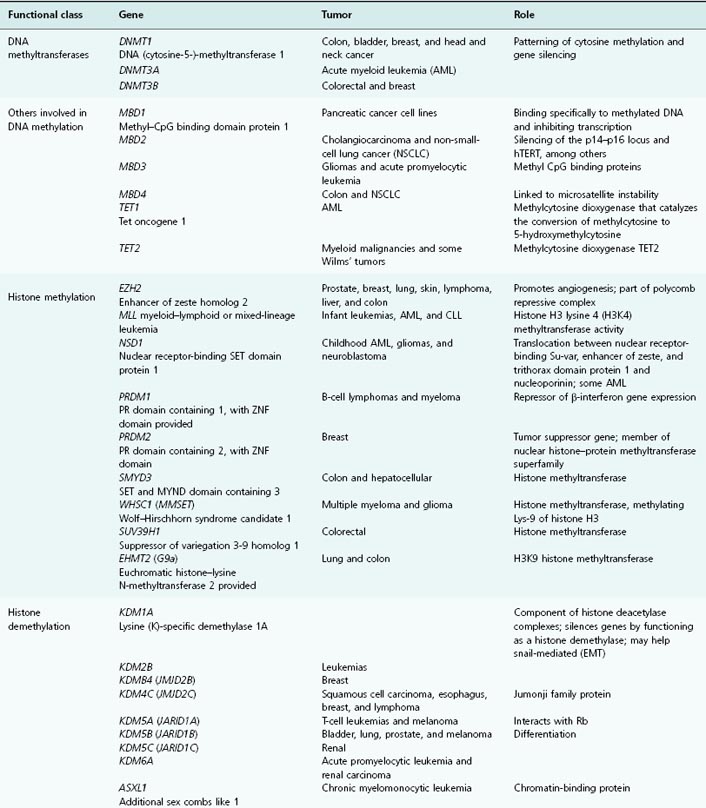
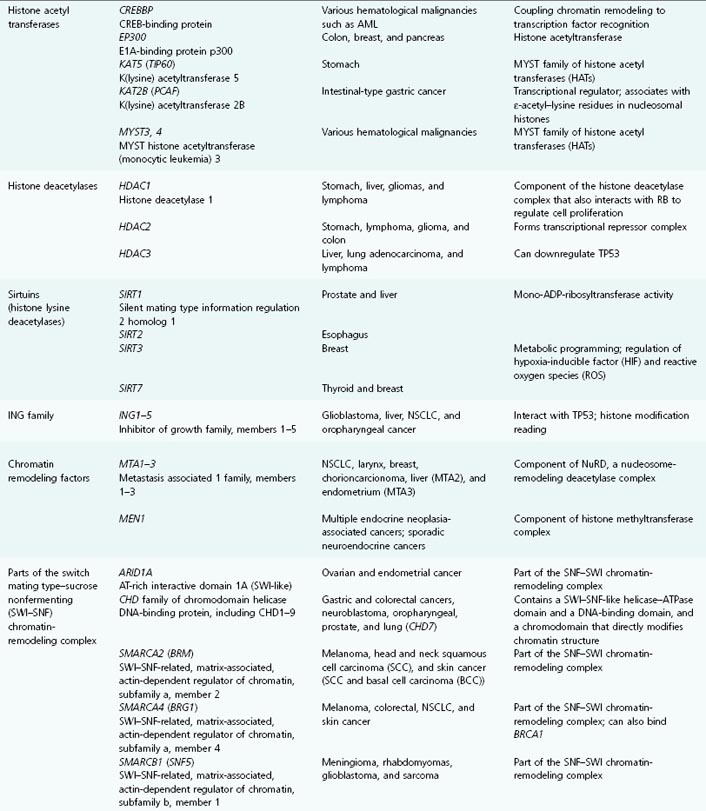
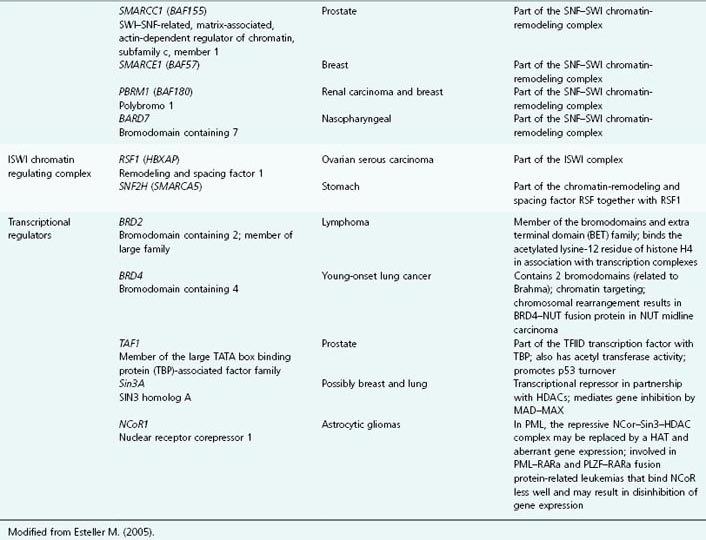
CpG Islands
Roll on, deep and dark blue ocean, roll. Ten thousand fleets sweep over thee in vain. Man marks the earth with ruin, but his control stops with the shore.
Lord Byron
CpG islands are areas of greatly increased density of a dinucleotide sequence, cytosine–phosphate diester–guanine, which can form regions of DNA several hundred to several thousand base pairs long. The human genome contains around 45 000 CpG islands (comprising a total of around 50 million CpG dinucleotides), mostly found at the 5′ ends of genes. They are widely accepted as unmethylated in normal somatic cells except for those on the inactive X chromosome and some associated with imprinted genes. This is in contrast to the majority of CpGs that lie outside of islands and are methylated in mammalian genomes. Around 60% of all human gene promoters contain CpG islands around the promoter regions, and these include housekeeping genes (essential for general cell functions) and many frequently expressed in a normal cell. Most CpG islands are unmethylated and can be either transcriptionally active or inactive depending on the balance of transcriptional regulators and histone modifications. As will be seen in this chapter, CpG islands are important sites for epigenetic regulation of gene expression, and are frequently aberrantly methylated in cancer cells. However, exactly how CpG-island promoters may become hypermethylated in cancer is not known. But recent papers have highlighted the potentially critical role of the enzyme TET1 in catalyzing hydroxylation of mC9, which together with hmC could regulate both DNA demethylation and gene expression. In this case, loss of TET1 would be predicted to result in promoter hypermethylation.
Epigenetics
Our deeds determine us, as much as we determine our deeds.
George Eliot
In many ways what is now referred to as epigenetics is not new science – it has long been recognized that traditional genetics theory, which implied a one-to-one relationship between genotype and phenotype, could not readily explain processes such as cell differentiation; here multiple, often very different cell phenotypes are produced, yet all bear ostensibly the identical genome. Thus, it was hypothesized that each undifferentiated cell underwent a crisis that determined its fate (a sort of cellular 11-plus), which was not inherent in its genes and was therefore, from the Greek, epigenetic – “in addition to” – the genetic information encoded in the DNA (see Box 11.2 for a historical overview). We now appreciate that the selective silencing of some genes and expression of others during development ultimately determine the phenotype of every cell, and that so-called epigenetic factors are key determinates of this. Epigenetics is one of the most exciting areas of modern biology, and particularly over the last few years we have developed a much clearer understanding of how this mechanism operates, and how epigenetic factors can control gene expression.
Changes to DNA and its associated proteins can alter gene expression without altering the DNA sequence. DNA is not found in isolation in the cell but is associated with proteins called histones to form a complex substance known as chromatin. Chemical modifications to the DNA or the histones alter the structure of the chromatin without changing the nucleotide sequence of the DNA. Such modifications are described as epigenetic. Changes to chromatin structure have a profound influence on gene expression: if the chromatin is condensed, the factors involved in gene expression cannot get to the DNA, and the genes will be switched off. Conversely, if the chromatin is in an “open” conformation, the genes can be expressed on demand for cellular activities. Unraveling these processes has been important as we now appreciate that in addition to the well-known role of DNA sequence changes (mutations), aberrant gene expression also results from more recently identified changes in gene silencing, due to epigenetic modifications.
Another useful metaphor, with which the reader will often be confronted, contends that genetic information provides a blueprint for manufacturing proteins necessary to create the organism, while the epigenetic information provides additional instructions on how, where, and when the genetic information is deployed. Epigenetic information is not contained within the DNA sequence itself, but can still determine mitotic inheritance of various characteristics as surely as modifications in the DNA sequence. Thus, epigenetic factors, which include DNA methylation and histone modifications, can dictate cell fate and gene expression patterns in the progeny after cell division and are important in the normal regulation of differentiation, aging, and senescence. Epigenetic factors can even turn environmental effects into heritable changes in cell phenotypes – which at face value challenges the central dogma of genetics. However, adult patterns of methylation are generally believed to be erased during early embryogenesis so that, in general terms, cells in a new organism are believed to start life with an epigenetically “clean slate.” Subsequently, during development and adult life, cells progressively acquire epigenetic “chalk marks” that they can pass on to their progeny. Importantly, all epigenetic modifications are not invariably predetermined during ontogeny but are influenced throughout life by genetic and environmental forces. Intriguingly, such epigenetic factors may also be the explanation for differences in phenotype observed between genetically identical twins.
Over the last couple of years, important studies have convincingly demonstrated examples whereby epigenetic modification can under some circumstances result in the translation of life experiences in a parent into inherited alterations in gene expression in the offspring. Numerous examples, relating nutritional deprivation or excess in a parent to subsequent risk of obesity and diabetes in the offspring, have been demonstrated. Much of this was perceived to be in utero conditioning. However, intriguingly, a recent paper in Nature in 2010 suggested that a paternal high-fat diet (HFD) resulted in development of β-cell dysfunction in female rat offspring with altered expression of multiple pancreatic islet genes at least in some cases by altered methylation. At least in this case, offspring are inheriting the life experience of the father.
Methylation of DNA
Musicians paint their pictures on silence. We provide the music, and you provide the silence.
Leopold Stokowski
The prototypic epigenetic modification of DNA in mammalian cells is the covalent addition of a methyl (CH3) group to the fifth position of cytosine within CpG dinucleotide islands, which can directly turn off gene expression (“silencing”) (Fig. 11.1). The methylation of DNA is achieved by three DNMTs, termed DNMT1, DNMT3A, and DNMT3B, though exactly how these are targeted to specific DNA regions is unclear. The other major class of epigenetic modification involves posttranslational modification of histones and chromatin remodeling (see the “Epigenetics and cancer” section).
Figure 11.1 Methylation can inhibit gene transcription. In the presence of CTCF, the gene is “insulated” from methylation; in this case, the gene is transcriptionally active as it will remain in the unmethylated state. Conversely, in heterochromatin, CpG islands in the promoter region are methylated, and these regions are transcriptionally inactive. Gene silencing following methylation is reinforced by deacetylation and interactions with repressors of transcription. In fact, DNMTs, which further mediate methylation, may be actively recruited to help maintain silencing. Methylation in turn enables the binding of a complex comprising the methyl cytosine-binding protein (MBP) and histone deacetylase (HDAC); some MBPs (MECP2 and MBD1/2) can also associate with transcriptional co-repressors such as SIN3, which directly bind to HDAC and contribute to gene silencing. HDAC promotes deacetylation of histones, which contributes to organization of nucleosomes and also, more generally, repression of transcription. In order for gene expression to take place, the HDAC–SIN3 complex is displaced and a transcription activator complex (transcription factor, histone acetyl transferase (HAT), and coactivator protein) can associate with promoter elements; HAT acetylates the histone-reversing effects of HDAC. In cancer, many genes may be inappropriately inhibited by methylation in the promoter region, and this is a frequent cause of loss of tumor suppressor activity during tumorigenesis.
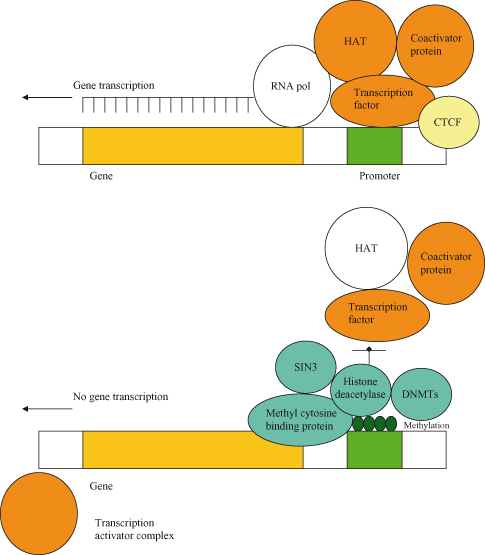
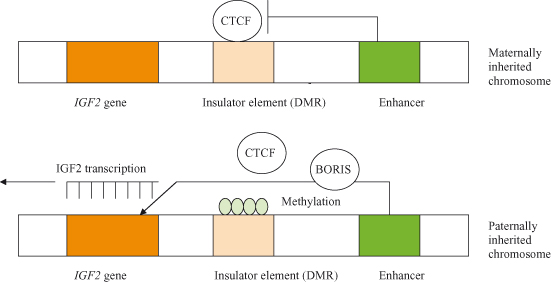
It is believed that DNA methylation may have evolved for silencing of repetitive elements, but has subsequently been adopted in order to effect transcriptional silencing in imprinting and X-chromosome inactivation. Imprinting, the phenomenon whereby expression of a gene may be silenced depending on whether it was inherited from the mother or the father, is thought to be due to differential methylation in maternal versus paternal genes (Fig. 11.2). Conversely, loss of imprinting refers to either the activation of normally silent imprinted genes or potentially the silencing of active imprinted genes, and is frequently observed in many different cancers. In particular, reactivation of the normally imprinted allele of the IGF2 gene is often seen in human cancers and is associated with resistance to apoptosis and tumor progression in animal models.
Figure 11.2 Imprinting of the gene for IGF2. On chromosomes inherited from the female, a protein called CTCF binds to an insulator preventing interaction between the enhancer and the IGF2 gene. IGF2 is therefore not expressed from the maternally inherited chromosome. Because of imprinting, the insulator on the male-derived chromosome is methylated; this inactivates the insulator by blocking the binding of the CTCF protein, and allows the enhancer to activate transcription of the IGF2 gene. It is speculated that CTCF is displaced by another protein, BORIS. The methylation patterns (imprints) on the chromosome, inherited by the zygote after fertilization, are maintained in subsequent generations by maintenance methyl transferases. After Alberts et al. (2002).
Around half of all genes have a CpG island in their promoter region, but most such CpG island–rich promoters are not methylated, irrespective of the expression state of the associated gene. This suggests that methylation of these promoters is not normally involved in the day-to-day regulation of gene expression in the large majority of cases. However, in areas where gene expression is silenced, such as the silenced allele of imprinted genes and the inactive X chromosome in females, promoter-associated CpG islands are methylated.
Methylation is required for silencing of genes, but the actual mechanisms responsible for establishing it still remain unclear. However, some of the consequences of CpG island methylation are now known and include binding of methylated DNA-specific binding proteins to CpG islands that then help recruit various histone-modifying enzymes responsible for restructuring the chromatin. DNA methylation by DNMTs modifies the actual DNA itself, can directly prevent gene expression by preventing transcription factors binding to promoters, and can additionally exert a more general effect by recruiting methyl-binding domain (MBD) proteins. These are associated with further enzymes called histone deacetylases (HDACs), which function to chemically modify histones and change chromatin structure. Histones may also become methylated on lysine residues, and this may contribute to gene silencing and imprinting; in fact, it is increasingly likely that the machinery controlling DNA and histone methylation is linked and collaborates in gene silencing. Thus, methylation of a CpG island alters expression of a gene in two ways, directly by interfering with the binding of specific transcription factors to promoters and indirectly by recruiting proteins such as MBD that associate with HDACs, which function to deacetylate histones and change chromatin structure.
Thus, epigenetic regulation depends on two overlapping processes:
- methylation of the DNA; and
- posttranslational modification of histones.
Acetylation of Histones and Other Posttranslational Modifications
As we must account for every idle word, so must we account for every idle silence.
Benjamin Franklin
As discussed earlier, histone acetylation plays an important role in the regulation of gene expression and together with methylation can influence the binding and activity of transcriptional activator complexes (Fig. 11.1). In fact, it has recently become clear that deacetylation of histones by HDAC is a major means by which methylation results in formation of heterochromatin and in the suppression of gene expression. Most of the human genome is packaged up as transcriptionally inactive densely packed heterochromatin, and this chromatin is heavily methylated. The remainder of the genome is transcriptionally active but still subject to various stimulatory and inhibitory processes that control gene expression on a day-to-day basis.
Studies of heterochromatin have helped unravel the complex mechanisms by which methylation of DNA culminates in silencing of gene expression. DNA in methylated regions is packaged into dense nucleosomes, which also contain deacetylated histones such as deacetylated H3 and also H4. Histone acetylation has a direct effect on the stability of nucleosomal arrays and on chromatin structure.
Histone acetylation is a dynamic process that is regulated by two groups of opposing enzymes, the histone acetyltransferases (HATs) and the histone deacetylases (HDACs) – see Fig. 11.3. Through these effects on histone (and also nonhistone proteins – see later), these enzymes play key roles in regulation of gene expression (see also the section on c-Myc in Chapter 6), chromosome segregation, and development. Moreover, their deregulation has been linked to cancer. The HATs catalyze the covalent addition of an acetyl group from acetyl coenzyme A to the N-terminal lysine residues of histones, whereas the HDACs remove such acetyl groups. Histone acetylation is a defining feature of transcriptionally active chromatin, and in the presence of appropriate transcriptional activators the gene will be expressed (note expression is still subject to regulatory factors). Conversely, the cardinal features of constitutive heterochromatin (transcriptionally inactive chromatin – genes not expressed) include deacetylation of histones as well as hypermethylation of DNA.
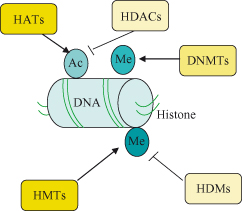
In keeping with this, inactive genes are associated with bound complexes containing HDACs that deacetylate histones, whereas active genes have strongly acetylated histones under the influence of HAT activity that reconfigure the chromatin to be open and accessible to the transcriptional activator complex. Histone deacetylation causes the condensation of chromatin, making it inaccessible to transcription factors, and the genes are therefore silenced (see Figs 11.1 and 11.3).
In fact, a number of other crucial posttranslational modifications of histones, in addition to acetylation, have been identified. These include methylation, phosphorylation, ubiquitination, sumoylation, glycosylation, and ADP ribosylation, all of which can help regulate the activity of many genes by modifying both core histones and nonhistone transcription factors. Also, we must not forget the importance of the variant histone yH2AX in the DNA damage response (see Chapter 10).
Recent studies have identified what has become known as a “methylation mark” that may help define and separate regions of transcriptionally active chromatin from transcriptionally inactive chromatin. These marks seem to involve methylation of lysine 9 in the tail of histone H3, which marks inactive and methylation of lysine 4 on histone H3 which marks transcriptionally active chromatin. The methylated lysine 9 appears to bind proteins required for maintaining a repressed state, but it remains to be shown how this leads to methylation of DNA. Possibilities include the facilitation of binding of DNMTs. What seems clear is that despite the complexity of epigenetic factors, methylation appears a dominant event over acetylation, as in cancer inhibiting HDAC alone does not reactivate aberrantly silenced genes and hypermethylated genes, whereas these same inhibitors can if cells are first treated with demethylating drugs.
The Histone Code
Since it became clear that the genome contains information in two forms, genetic and epigenetic, research efforts have been directed at trying to crack the “histone code” which is in many respects analogous to the DNA code which was unraveled many years ago.
As you will have gathered, histones not only are there to pack away the DNA, but also are pivotal regulators of chromatin structure and function, at least in part because they can integrate a variety of regulatory processes which operate through various posttranslational modifications of the histone tails. The “histone code” hypothesis, which postulates that these covalent histone modifications regulate gene transcription, was first proposed by Strahl and colleagues more than a decade ago. According to this hypothesis, histone modifications, such as lysine methylation, are “read” by specific binding proteins that enhance or suppress transcription depending on the site that has been “marked.” In some ways, this histone code is complementary to the DNA code and determines how and when specific genes are transcribed. In addition to methylation and acetylation, other modifications used in the histone code include phosphorylation, ubiquitination, sumoylation, and ADP ribosylation within gene regulatory regions. Although the consequences of such modifications are not well known, they likely include chromatin restructuring and controlling the docking or function of transcription factors or other histone-modifying enzymes. This model offers an explanation for how modest single or small numbers of histone modifications might regulate chromatin functions, particularly gene transcription. Some progress has now been made in decoding histone modifications. For example, gene expression can be activated by monomethylation of lysines (K) at position 20 and 5 of histones H4 and H2B to form H4K20 and H2BK5 respectively or by trimethylation of lysines 4, 36, and 79 on H3 to form H3K4me3, H3K36me3, and H3K79me respectively. Genes are also activated by acetylation of H3K9 and H3K14. Conversely, gene expression is repressed by trimethylation of H3K9 and H3K27.
In many cancers, trimethylation of H4K20 and acetylation of H4K16 are reduced, and alterations in many of the enzymes responsible for histone posttranslational modifications and chromatin structuring have been demonstrated in a wide range of cancers and pre-malignant conditions. Thus, several HDACs, HMTs and HDMs, lysine acetyltransferases, sirtuins, and the JARID-1 family have been shown to be abnormally expressed in a variety of different cancers. See Table 11.1 for the lengthy list of genes encoding epigenetic regulatory proteins that are aberrantly expressed (up or down) or mutated in human cancers.
Epigenetic Regulation of Gene Expression
Chromatin structure, nucleosome modeling, and promoter DNA methylation are amongst the most important determining factors in gene expression. We will now focus more on exactly how chromatin modification is recognized and acted upon by the transcriptional machinery, thereby turning histone coding into altered gene expression. The enzymes responsible for the posttranslational modification of nucleosomal histones, such as the DNMTs, have been described already (and are listed in Table 11.1, alongside the cancers that may result from their aberrant expression or function).
Histone modifications act by recruiting a number of modification-specific binding proteins, such as 14-3-3, that facilitate transcriptional activation. Gene expression is influenced by signaling pathways in several ways, including by histone modification, which is of relevance here, as exemplified by phosphorylation of histone H3. Expression of several key immediate-early (IE) genes is regulated in part by H3 phosphorylation at S10 (H3S10ph) at promoters and coding regions when MAPK signaling is activated. This might appear simple, but given the fact that a large number of kinases can directly phosphorylate H3, such as MSK1/2, PIM1, RSK2, and IKKα, it is distinctly possible that this might be a central regulatory node in the signaling-related activation of gene expression. H3 S10 phosphorylation is linked to acetylation of adjacent lysine residues (K9 or K14) suggesting rewriting of the histone code, which in turn can promote binding of various 14-3-3 proteins required for induction of gene transcription of several IE and HDAC genes.
How this actually works is exemplified by the FOSL1 gene, where 14-3-3 binds to H3S10ph at the enhancer and then recruits a cohort of regulators including HAT, BRD4, males absent on the first (MOF), and the transcription elongation factor b (P-TEFb), whilst H3 S10 phosphorylation also restarts the preinitiated but paused RNA polymerase II. H3 S28 phosphorylation may also facilitate binding of 14-3-3 and has been observed at nucleosomes at IE gene promoters. A recent study by Lau and Cheung has shown that S28 phosphorylation, which induces a methyl–acetylation switch on an adjacent K27 residue, is important in the direct activation of the IE gene c-fos and the polycomb-silenced α-globin gene. The authors also suggest how histone coding differences might operate in different contexts; H3 S10 phosphorylation might facilitate transcriptional elongation of genes that are regulated by polymerase pausing, whereas H3 S28 phosphorylation might directly initiate transcription.
At least 90% of all human genes are subject to alternative splicing, which will be discussed in more detail later, but given the key role in determining the types of protein produced by a gene, it is important to consider how this is regulated. Chromatin as the template for nuclear transcription can influence splicing choices by altering structure and the way in which the histone code is interpreted. It is now known that nucleosomes are often located at exon–intron boundaries and are susceptible to specific histone modifications which can in turn regulate alternative splicing.
The mSin3A corepressor is a core component of a large multiprotein corepressor complex that links HDACs with chromatin targeting subunits such as Pf1 and MRG15. In man, an Rpd3S–Sin3S corepressor complex represses aberrant gene transcription from cryptic transcription initiation sites and blocks progression of RNA polymerase II in actively transcribed genes. A recent computational analysis by Ron DePinho and colleagues has confirmed the wide range of genes regulated by this means. Thus, several nodal points by which mSin3A influences gene expression have been identified, including the Myc–Mad, E2F, and p53 transcriptional networks.
The tumor suppressor RB is also a key player in the assembly of constitutive heterochromatin. As we have seen in Chapters 4 and 7, RB represses genes required for entry into the S phase and progression of the mitotic cell cycle, an action at least in part long appreciated to involve interaction with HDAC at the promoter regions of various genes activated by E2F family transcription factors. But recent studies, some from our coauthor Maria Blasco, suggest that RB may function as a global suppressor of gene expression and that moreover loss of RB could result in a generalized loss of repressive chromatin and reactivation of gene expression. Importantly, epigenetic factors may be a general way by which proteins and genes communicate with each other, and in particular histone acetylation is now known to contribute to the day-to-day regulation of gene expression. Thus, inhibition of gene expression by transcription factors such as c-MYC involves the recruitment of histone-modifying co-repressor complexes containing HDAC and mSIN3A (or, in other such complexes, N-CoR–SMRT), which can promote deacetylation of lysines in histone H4 tails. In many cases, it now appears that for transcriptional activation of multiple genes, epigenetic factors (possibly generally involving inhibitory complexes of RB and HDAC, among others) must first be overcome. In other words, the role of epigenetic factors in controlling gene expression is extending way beyond the previous notion of such factors predominantly mediating permanent or near-permanent inactivation of genes in development. This is graphically illustrated by the means by which some genes are inhibited and others activated during regulation of the G1–S transition in the mitotic cell cycle, discussed in Chapter 4. To remove the brakes from the cell cycle, c-MYC, in partnership with MIZ1, inhibits expression of genes such as the cyclin-dependent kinase inhibitor (CKI) p21CIP1 by recruiting histone-modifying complexes containing mSIN3A and HDAC to the promoter regions and concurrently promotes expression of genes (cyclin–CDKs, etc.) in partnership with a different protein MAX. The net effect is hyperphosphorylation of the RB protein, which in turn displaces both RB and associated histone-modifying enzymes such as HDAC from the various promoters essential in order for E2F family transcription factors to drive expression of genes needed for the S phase.
Epigenetics and Cancer
Epigenetic changes are key factors in several diseases, in particular cancer – and their importance is underscored by the devotion of a large part of this chapter to the topic. As discussed, the major forms of epigenetic modification that have been associated with cancer cells are aberrant DNA methylation of CpG islands located in gene promoter regions (including loss of imprinting of genes) and changes in chromatin conformation involving histone deacetylation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


