- The immune system developed as a means for an organism to distinguish foreign invaders from normal host tissues.
- The tumor cell evolves from normal tissues that do not usually provoke an immune response. But through the expression of mutated products or the breaching of normal tissue barriers leading to an inflammatory milieu, the tumor cell may elicit an immune response.
- The immune response to a tumor cell initially activates early immune cells (the innate response) and then immune cells involved in antigen-specific recognition (the adaptive response).
- However, tumor cells can evolve and evade the immune response, leading to the clinical appearance of cancer.
- By dissecting the manner in which the immune response is activated, how immune cells recognize tumor cells, and the factors involved in augmenting or suppressing a tumor-specific immune response, effective immunotherapeutic strategies for cancer can be developed.
Introduction
The hypothesis that the immune system plays a role in the anti-tumor response and can be manipulated for the treatment of cancer was advanced as early as the 1890s, when William Coley used bacterial extracts to treat patients with sarcoma. It was thought that the ensuing inflammatory response successfully induced regression of large tumors by activating the immune system and inducing immune cells to attack the tumor. Since then, nonspecific forms of immunotherapy have been used with varying degrees of success for the treatment of a limited number of malignancies – BCG adjuvant for superficial bladder cancer, high-dose interleukin 2 (IL-2) for metastatic melanoma, and donor lymphocyte infusions for leukemic relapse after allogeneic stem cell transplant. However, these strategies were often accompanied by serious and potentially life-threatening toxicities. Advances in immunology, in the understanding of the requirements for T-cell activation and tolerance, and in the development of novel technologies to analyze and augment immune response now provide tumor immunologists with the opportunity to translate more broadly applicable principles in immunology to the practice of treating cancer patients in a more specific and effective manner.
In the last 5 years, several advances into the clinical arena have led to mainstream acceptance of immunotherapy as a rational treatment modality, led by reproducible clinical responses in the field of adoptive cellular therapy, with phase II and randomized phase III studies confirming the anti-tumor efficacy of vaccine therapy and the checkpoint inhibitor, anti-CTLA4.
This chapter on tumor immunology begins with a description of the endogenous immune response, followed by a discussion of the components involved, including T cells, dendritic cells, and tumor antigens, and finally a summary of immunotherapeutic strategies arising from an understanding of the anti-tumor immune response.
Endogenous Immune Response
The endogenous immune response is composed of two phases – the innate response, which provides the initial line of defense against tumors, and adaptive response, which provides longer lasting, antigen-specific immunity. These events are usually precipitated in response to a “danger signal” in the host that can be characterized by the introduction of foreign antigens or the disruption of the normal microenvironment by an invading tumor (see Box 13.1). As a first step, natural killer (NK) cells, representing the “rapid response” component of innate immunity, release proinflammatory cytokines such as interferon-γ leading to a cascade of soluble factors that include chemokines to recruit and activate macrophages and antigen-presenting cells (APC) such as dendritic cells (Fig. 13.1). Macrophages kill tumor directly through the release of lysosomal enzymes, reactive oxygen intermediates, and nitric oxide. Dendritic cells collect tumor fragments and tumor proteins and migrate to draining lymph nodes, where they process and present these proteins to antigen-specific CD4 and CD8 T cells, which lead to initiation of an adaptive immune response (Fig. 13.2). NK cells that can also mediate direct tumor cytotoxicity provide some degree of initial protection, but it is the adaptive response characterized by expanding populations of antigen-specific T cells and their differentiation into memory cells that provide the greatest potential for long-term immunoprotection against cancer. Naïve T cells, when they first encounter antigen (also known as priming), undergo a process of differentiation that leads to an effector population mediating tumor killing (see Box 13.2). A fraction of these cells persist as memory T cells. Memory T cells are T cells “at the ready,” requiring a much shorter time to become fully activated and expand than naïve T cells. This population of memory antigen-specific T cells provides for a more robust and rapid response when antigens reappear.
 , leading to its differentiation into an effector T cell and clonal expansion. Effector cells possess the ability to release cytokines and toxic granules (e.g. perforin and granzyme B), and mediate tumor cell killing
, leading to its differentiation into an effector T cell and clonal expansion. Effector cells possess the ability to release cytokines and toxic granules (e.g. perforin and granzyme B), and mediate tumor cell killing  . Following an effector response, the majority of antigen-specific T cells undergo apoptosis
. Following an effector response, the majority of antigen-specific T cells undergo apoptosis  , but a small fraction develop into memory T cells
, but a small fraction develop into memory T cells  that can survive in a quiescent state for many years after the antigen has been eliminated; these are found to reside in lymphoid and mucosal tissues
that can survive in a quiescent state for many years after the antigen has been eliminated; these are found to reside in lymphoid and mucosal tissues  . Upon antigen re-exposure
. Upon antigen re-exposure  , these memory cells are quickly called into action, having the capacity to be rapidly activated and to expand
, these memory cells are quickly called into action, having the capacity to be rapidly activated and to expand 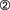 and kill tumor
and kill tumor  .
.
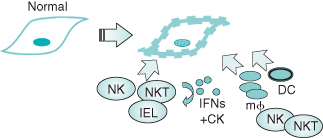
Figure 13.1 Innate anti-tumor immunity. See text for details. The development of tumor cells results in a proinflammatory milieu and activation of NK, NKT and IEL (intra-epithelial) cells. Release of cytokines and chemokines leads to recruitment of macrophages and additional innate effectors which provide an initial line of defense, and dendritic cells which initiate the adaptive immune response.
Cancer Immunosurveillance
That these defense mechanisms provide an immunologic basis for suppression of tumor cells by the endogenous immune response was hypothesized more than 40 years ago and has recently been supported by a number of murine models and clinical observations. In 1957, Burnet and Thomas postulated that “small accumulations of tumor cells may develop … and provoke an effective immunological reaction with regression of the tumor and no clinical hint of its existence.”
Figure 13.2 Adaptive anti-tumor immunity. Dendritic cells take up tumor antigen, and process and present antigen to CD4 and CD8 T cells in draining lymph nodes. Presentation of tumor antigen leads to CD4 and CD8 T-cell activation (cf. Fig. 13.3 for details). Activated CD4 and CD8 T cells traffic to tumor sites where they mediate an antigen-specific effector response through the release of cytotoxic granules, Fas–FasL interaction, and the recruitment of secondary effectors (cf. Fig. 13.4 for details).
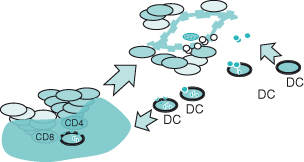
However, initial studies evaluating the incidence of chemically induced or spontaneous tumors in immunosuppressed mice treated by thymectomy or anti-lymphocyte sera led to conflicting results. Malignancies that did develop in immunodeficient mice were often virally induced or represented by lymphomas attributed to chronic antigenic stimulation from infectious agents. When athymic nude mice were evaluated for the development of spontaneous or chemically induced tumors, there was no increase in tumor incidence when compared to immunocompetent mice. Although not known at the time, athymic nude mice do retain a functional immune system composed of a small population of T cells and thymic-independent populations of NK cells. In the 1990s, mice were engineered with deficiencies in lymphocyte-specific recombinase, which was responsible for antigen receptor rearrangement (RAG-2 −/− mice). Such mice, which possessed no NK, T, and B cells, were found to develop sarcomas more frequently and with a shorter latency period than wild-type mice.
Interferon-γ was also shown to be protective against the growth of spontaneous and chemically induced tumors, and pivotal experiments using mice deficient for the interferon-γ receptor, or its signaling molecule, STAT1, demonstrated an increased incidence of chemically induced tumors that was over 10 times greater than that of wild-type controls. Mice deficient in both RAG-2 and STAT1 exhibited, in addition to inducible sarcomas, spontaneous intestinal and mammary cancers. Further, mice lacking perforin, which is released within cytolytic granules upon lymphocyte stimulation to effect tumor cell killing, were found to be more susceptible to sarcomas and spontaneous lymphomas. Interestingly, chemically induced tumors developing in immunodeficient (RAG-2 −/−) mice, when injected into wild-type, immunocompetent mice, were found to be more readily rejected than those tumors developing in immunocompetent mice. These results suggested that the innate immune response in immunologically intact hosts may be instrumental in sculpting the immunogenic phenotype of tumors and may, over time, result in immunoselection that leads to tumor immune escape.
Cancer and Immunosuppression
The role of the immune system in suppressing the development and progression of human malignancies was suggested initially in observational studies evaluating infiltrating or intratumoral lymphocytes in melanoma biopsy specimens. A correlation between the extent of mononuclear infiltration and prognosis has been extended for breast, colon, and most recently ovarian cancer in which patients whose tumors contained T cells experienced a 6–8-fold greater 5-year overall survival rate than those whose tumors contained no T cells. Conversely, patients who are chronically and/or severely immunosuppressed, as a result of stem cell ablation, HIV, or induced immunosuppression following transplantation, experience a higher overall incidence of malignancy than the general population. Although melanomas and occasionally colon, lung, and bladder cancers are observed, most tumors that develop are virally related, for example Epstein–Barr virus (EBV) post-transplant lymphoproliferative disease progressing to lymphoma, Kaposi sarcoma, or human papilloma virus (HPV)–associated malignancies. This may be consequences of a shorter latency period for viral-associated malignancies and the shorter life span of immunosuppressed patients, which preclude the appearance of longer developing malignancies. In some cases, reversal of immunosuppression has resulted in dramatic responses.
Effector Cells in Tumor Immunity
Effectors of adaptive immunity can be ascribed to “humoral” and “cellular” arms, represented respectively by B cells that mediate effects through the production of antibodies, and T cells that interact directly with target cells through the T-cell receptor. In humoral immunity, antibodies binding surface proteins on tumor cells can kill via complement activation or by bridging targets with cytotoxic cells through a process known as antibody-dependent cell-mediated cytotoxicity (ADCC). In this process, the Fc portion of antibody couples with receptors on macrophages or NK cells, which then effect cell killing. Although antibodies are highly effective in vitro, convincing evidence that antibody responses elicited in vivo play a critical role in anti-tumor immunity is weak. However, the significance of humoral responses with respect to tumor immunity has been supported by the identification of serum antibodies to potentially immunogenic tumor antigens (see the “SEREX” section of this chapter) and the successful therapy of patients using monoclonal antibodies (see the “Antibody therapy of cancer” section).
Dendritic Cells
Dendritic cells are specialized or “professional” antigen-presenting cells that are activated during the innate immune response and are uniquely equipped to take up and present antigen to effector cells of the adaptive immune system – the antigen-specific CD4 and CD8 T cells. Dendritic cells are so-named because of pseudopods or “dendrites,” which are processes that extend from the cell to facilitate antigen presentation. In vivo, the induction of an anti-tumor immune response may occur by tumor cells presenting antigen directly to T cells, but it is believed that a more common and robust pathway for tumor-specific T-cell activation in vivo is by cross-presentation. This is a process by which antigens released by necrotic, dying, or apoptotic tumors are taken up by dendritic cells and re-presented to T cells under more favorable stimulatory conditions in the tumor-draining lymph node.
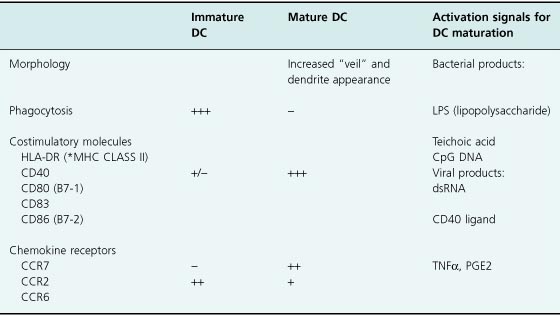
Dendritic cells can be characterized in their immature or mature forms based on contrasting surface and functional phenotypes (Table 13.1). In their immature form, dendritic cells are well equipped to capture antigens through surface receptors such as the C-type lectins (e.g. DEC-205 and mannose receptors), αvβ5 integrins, or CD36 for internal processing and presentation. Dendritic cell activation via “danger signals” mediated through some of these receptors and other surface receptors (e.g. the Toll-like receptors) leads to DC maturation in the presence of bacterial or viral products, TNF-α, or prostaglandins. It is also thought that in addition to DC activation by these receptors, cooperation of CD4 helper T cells is required to “license” dendritic cells with the capacity to stimulate CD8 T cells through interaction of the CD40 ligand on CD4 T cells with CD40 on dendritic cells. Upon maturation, further antigen uptake by dendritic cells is downregulated, and, in preparation for optimizing T-cell activation, dendritic cells upregulate surface expression of the T-cell costimulatory molecules (CD80, CD83, and CD86), and secrete cytokines such as IL-7 and IL-12 which facilitate full T-cell activation. In the case of tumor immunity, dendritic cells circulate through the blood and accumulate at tissue sites in response to chemokines arising from the site of tumor necrosis or inflammation. As immature dendritic cells, antigen is collected and processed for presentation on the surface in the context of major histocompatibility complex (MHC) molecules. Following maturation and upregulation of surface costimulatory molecules, lymphokines, and the chemokine receptor CCR7, dendritic cells traffic to lymph nodes where T-cell activation can occur.
Table 13.1 Immature versus mature dendritic cells – surface and functional phenotypic differences
T Cells
Antigen Presentation and T-Cell Stimulation
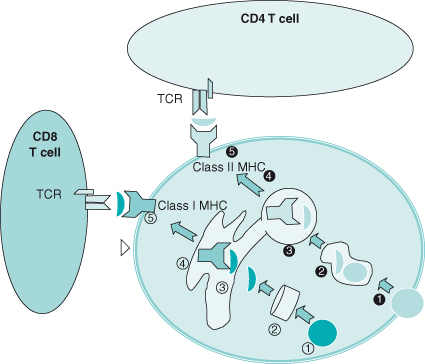
In contrast to B cells, which provide “humoral” immunity through the production of soluble antibodies, T cells mediate “cellular” immunity by interacting directly with their target cell. T cells achieve specificity for cells expressing the target antigen through the surface T-cell receptor which recognizes fragments of antigen (usually peptide fragments) presented by the MHC through either a Class I or Class II processing pathway (Fig. 13.3). Since proper antigen processing and presentation by the MHC are critical to antigen-specific immunity, a brief description of the MHC complex is presented.
Figure 13.3 Antigen presentation. In the Class I pathway (  ), cytosolic proteins are processed by proteasomes
), cytosolic proteins are processed by proteasomes  into peptide fragments, transported through the endoplasmic reticulum (ER,
into peptide fragments, transported through the endoplasmic reticulum (ER,  ), complexed with Class I MHC
), complexed with Class I MHC  and β2-microglobulin, and brought to the surface
and β2-microglobulin, and brought to the surface  , where they are presented to CD8 T cells.
, where they are presented to CD8 T cells.
In the Class II pathway, extracellular protein antigens are endocytosed  , degraded into peptide fragments
, degraded into peptide fragments  , combined with MHC
, combined with MHC  , and presented to the surface
, and presented to the surface  where they are presented to CD4 T cells
where they are presented to CD4 T cells  .
.
The MHC is encoded by highly polymorphic genes that cluster on chromosome 6 in humans and are co-dominantly expressed. Human MHC molecules are called human leukocyte antigens (HLA) and are divided into Class I and Class II HLA or MHC, which present peptides to CD8 and CD4 T cells, respectively. For the most part, Class I MHC molecules are represented in humans by the HLA-A, -B, and -C families of alleles, and Class II MHC by the HLA-DR, -DQ, and -DP families. The Class I MHC complex is composed of three parts: the MHC-encoded heavy α chain, a non-MHC-encoded β2-microglobulin chain, and an 8-11-mer peptide sitting in a groove formed by the polymorphic region of the α chain. The Class II MHC complex is composed of three parts, the polymorphic α and β chains and a 10-30+ mer peptide sitting in a groove formed by the two chains. These parts are assembled in cytosolic compartments together with peptides derived from tumor proteins that have undergone proteosome-mediated cleavage to peptides of the appropriate length. The peptide–MHC complex is then transported to the surface, where the immunogenic peptide sitting in the MHC groove is presented to the T-cell receptor. The T cell recognizes the peptide only in the context of the MHC complex; therefore, mutations affecting any component of the antigen0presenting machinery can abrogate specific T-cell recognition and killing of tumor cells.
Costimulatory and Inhibitory T-Cell Signals
In addition to the interaction of the T-cell receptor (TCR) with the peptide–MHC complex, the activation of T cells can be modulated by the engagement of surface costimulatory or accessory molecules by their respective ligands on antigen-presenting cells (see Box 13.3). The most prominent of these is the signals provided by CD28 upon binding to B7-1 (CD80) or B7-2 (CD86) on APC. B7–CD28 interaction mediates signals that can fully activate an antigen-driven T-cell response, enhance T-cell survival by upregulation of antiapoptotic proteins such as BCL-xL, and drive proliferation. Absence of B7 has been associated with T-cell energy, while engineered expression of B7 in potentially immunogenic tumor cells can induce tumor rejection in murine models. While B7–CD28 interaction appears to be critical to the generation or priming of an effective anti-tumor response, it does not influence the effector or killing phase of T cells. Hence, T cells generated with a B7-transduced tumor vaccine can eradicate B7-negative tumor. Other costimulatory molecules that deliver a positive signal to T cells include ICOS (inducible costimulator), OX40, 4-1BB, and other B7 family members (e.g. B7–H3). Accessory or adhesion molecules such as ICAM-1 and LFA-1 are also critical to T-cell recognition. These molecules converge in and reinforce the TCR–peptide–MHC synapse by forming in aggregate with other molecules, a supramolecular activation complex to facilitate delivery of a longer lasting, more potent T–cell signal.
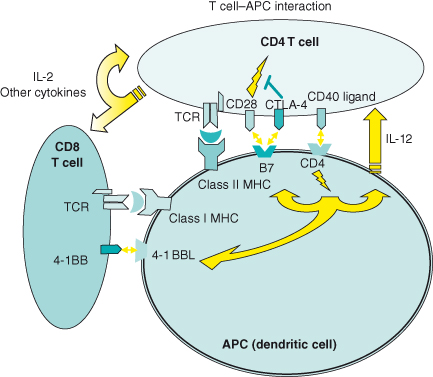
Cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) delivers a negative regulatory signal to activated T cells and competes with CD28 for binding to B7 on target cells (see Box 13.4). Since the overall effect is to block cell-cycle progression, CTLA4 is considered a checkpoint inhibitor. CTLA-4 is present predominantly as an intracellular protein and appears on the surface as an inducible receptor with a greater affinity for B7 than CD28; however, in contrast to CD28, its surface expression is nonconstitutive and relatively short-lived. CTLA-4 is believed to provide an immunologic “brake” to prevent overly robust and potentially damaging overstimulation by suppressing T-cell proliferation through IL-2 inhibition and downregulation of cell-cycle activity. CTLA-4-deficient mice develop splenomegaly and a lymphoproliferative pathology. Since many tumor target antigens are also normal self-proteins, eliminating CTLA-4 inhibition may provide a means of breaking tolerance to self-antigens and augment an otherwise muted T-cell response to tumor. Administration of anti-CTLA-4 antibody in some murine models results in organ autoimmunity but can also lead to rejection of previously non-immunogenic tumors. In clinical trials, administration of anti-CTLA4 antibody has produced signs of autoimmune toxicity as well as tumor regression in individuals receiving a tumor-specific vaccine.
 )
) ):
): The vaccine reagent may be:
The vaccine reagent may be:- Whole tumor cells, cell lysates, or tumor cell lines that are genetically modified (e.g. with GM-CSF to augment immunogenicity) can be used as vaccines.
- Peptides or DNA encoding the antigen of interest may be used directly as vaccine reagents.
- Dendritic cells charged with peptide or protein or transfected with recombinant vectors encoding the antigen of interest represent a potentially greater immunogen.
- Adjuvant (e.g. BCG) or adjuvant cytokines (e.g. IL-12) may be added to enhance immunoreactivity.
 The route of administration may be:
The route of administration may be:- Intradermal.
- Intramuscular (e.g. plasmid DNA).
- Intralymphatic.
 Augmentation of antigen-specific immunity may require several booster immunizations after initial priming; maximal levels may not be achieved until several weeks after vaccination.
Augmentation of antigen-specific immunity may require several booster immunizations after initial priming; maximal levels may not be achieved until several weeks after vaccination. ):
): Leukapheresis:
Leukapheresis:- Is a procedure that collects white blood cells from patients and provides peripheral blood mononuclear cells as a source of dendritic cells and T cells for in vitro stimulation cultures.
 Isolation of antigen-specific T cells in vitro.
Isolation of antigen-specific T cells in vitro.- Requires a source of stimulator and responder cells. Stimulator cells can be dendritic cells pulsed with peptide or engineered to express antigen following transfection or viral infection with recombinant vectors encoding the target gene of interest. Responder cells can be PBMC enriched for CD4 or CD8 T cells. Iterative restimulation cycles are performed to augment the fraction of antigen-specific T cells in vitro.
 T cells demonstrating specificity for the target antigen are identified and expanded in flasks or culture bags to several billion using a combination of feeder cells, TCR stimulation, and cytokines.
T cells demonstrating specificity for the target antigen are identified and expanded in flasks or culture bags to several billion using a combination of feeder cells, TCR stimulation, and cytokines. Expanded T cells are adoptively transferred into patients; high frequencies of in vitro–generated, antigen-specific T cells may be achieved in vivo with multiple infusions.
Expanded T cells are adoptively transferred into patients; high frequencies of in vitro–generated, antigen-specific T cells may be achieved in vivo with multiple infusions.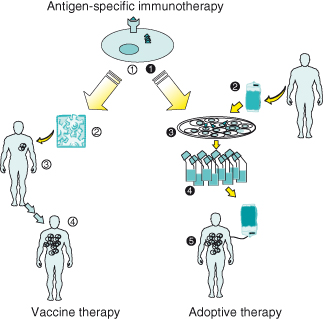
Programmed death-1 (PD-1) is another checkpoint inhibitor responsible for maintaining the balance between activation and suppression of T-cell responses. Its ligands PD-L1 (or B7-H1) and PD-L2 (B7-H2) are expressed on antigen-presenting cells, nonhematopoietic cells, and tumor cells. Bidirectional signaling between B7-1 and PD-L1 has also been observed, leading to an overall inhibitory effect. Blocking of PD-1–PD-L1 interactions has led to restoration of proliferative and cytokine capacity.
PD-L1 is aberrantly expressed on tumor cells and is a poor prognostic feature in patients with many solid tumor malignances. A PD-L1 blockade appears to enhance concomitant immunotherapies in animal models of vaccine and adoptive therapy. Anti-PD-1 therapy can also lead to reversal of effector cell exhaustion and increased cytotoxic T-lymphocyte (CTL) resistance to Treg-mediated inhibition.
T Lymphocytes
T cells can generally be divided into helper CD4 T cells and cytotoxic or killer CD8 T cells. Helper CD4 T cells recognize antigen in the context of MHC Class II and can be further differentiated into Th1 and Th2 subsets on the basis of distinct cytokine and receptor profiles. Th1 CD4 T cells produce IL-2 and interferon-γ, and express IL-12 and IL-18 receptors and regulate T-cell immunity, while Th2 T cells produce IL-14, IL-15, and IL-13, and regulate B-cell immunity. It is believed that a Th1-type response would be beneficial in anti-tumor immunity since it mobilizes a T-cell-mediated response.
Cytotoxic CD8 T cells recognize antigen in the context of MHC Class I and, when activated, release perforin and toxic granules that mediate direct cell killing by punching holes in the cell membrane to facilitate entry of enzymatic packets (granzymes A and B). Although most studies have weighed in on a greater role for the cytotoxic CD8+ T lymphocyte (CTL) in tumor eradication, the helper CD4 T lymphocyte has also been shown to be a vital component in the induction and maintenance of a competent anti-tumor immune response. Not only have tumor antigen-specific responses been identified for CD4 T cells, but also the presence of CD4 T cells may be required for CD8+ CTL responsiveness. Acting in concert, both CD4 and CD8 T cells provide for synergistic mechanisms of tumor killing. CD8 T cells kill tumor cells through the release of perforin and granzymes A and B, or through engagement of the death receptor, Fas, through Fas ligand (FasL) expressed on activated T cells. FasL–Fas interaction leads to a form of cell death known as apoptosis. In contrast to necrosis or death due to cell injury, apoptosis or programmed cell death involves a stepwise cascade of events initiated by receptor engagement at the cell surface (in this case, Fas), leading to DNA fragmentation. CD4 T cells can kill tumor cells directly by FasL–Fas engagement, as well as through indirect mechanisms that involve the recruitment of nonspecific effectors such as macrophages and eosinophils, which can act even on MHC-negative tumors (Fig. 13.4).
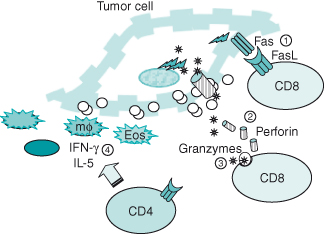
Figure 13.4 Effector mechanisms of T cells. Activated CD8 T cells deliver a “death” signal to tumor cells through Fas ligand–Fas interaction  . CD8 T cells may also kill tumor cells directly through perforin and granzymes released upon engagement of the T-cell receptor
. CD8 T cells may also kill tumor cells directly through perforin and granzymes released upon engagement of the T-cell receptor  . Perforin exocytosed in CTL granules forms pores in the tumor cell membrane. Granzymes enter tumor cells through pores and induce tumor cell death
. Perforin exocytosed in CTL granules forms pores in the tumor cell membrane. Granzymes enter tumor cells through pores and induce tumor cell death  . CD4 T cells can mediate tumor death through Fas interaction. Activated CD4 T cells may also mediate cytotoxicity indirectly through the release interferon-gamma and IL-5 to recruit tumoricidal macrophages (mϕ) and eosinophils (Eos)
. CD4 T cells can mediate tumor death through Fas interaction. Activated CD4 T cells may also mediate cytotoxicity indirectly through the release interferon-gamma and IL-5 to recruit tumoricidal macrophages (mϕ) and eosinophils (Eos)  .
.
Natural Killer Cells
NK cells are activated during the innate response by the inflammatory milieu that is established by invading tumor cells. These effector cells are not antigen-specific and do not express a TCR but do kill tumor through killer activating receptors (KARs) expressed on their surface. Engagement of KARs with tumor-derived ligands such as MICA and MICB, which are upregulated in infected or “stressed” cells such as tumor cells, leads to NK cell activation and tumor cell death. NK cells also engage self-MHC Class I molecules on target cells through inhibitory receptors (killer inhibitory receptors (KIRs)), perhaps as a means of preventing autoreactivity. The loss of MHC expression on tumor cells, a process which can develop during carcinogenesis and immunoselection, lends itself to preferential NK cell activation. The contribution of NK cells to the endogenous anti-tumor response in vivo may be best exemplified in athymic nude mice which have no T cells, but retain a population of functional NK cells which appears to be sufficient to mediate tumor resistance. In humans, NK-type cells can be expanded in vitro with high doses of IL-2 for adoptive transfer; however, in this setting, their efficacy is less well defined and treatment is often accompanied by serious toxicity. The in vivo augmentation of NK-type cells may also be one mechanism by which high-dose IL-2 therapy has shown some clinical effect in the treatment of patients with metastatic melanoma or renal cancer.
Regulatory T Cells
A population of T cells with regulatory properties that control autoimmune and anti-tumor responses was postulated as early as 1975; however, convincing evidence for their existence has been elusive. Recently, a population of CD4+ CD25+ T cells that possess immunosuppressive function has been identified. This discovery has led to a renewed understanding of the role of regulatory cells. CD4+ regulatory T cells (Tregs) are represented by two subsets – naturally occurring Tregs representing 5–10% of peripheral T cells, and induced Tregs that develop from conventional CD4+ CD25− T cells. Naturally occurring Tregs mediate their suppressive properties through cell-to-cell contact by an unknown mechanism. Although activation is dependent on TCR engagement, their suppressive effects are nonspecific. They are known to express glucocorticoid-induced TNF receptor (GITR) and CTLA-4, a known T-cell inhibitor of T-cell costimulation. However, the role of CTLA4 and GITR in mediating the suppressive effects of naturally occurring Tregs is not well defined. Induced or adaptive Tregs can be generated from conventional CD4+ CD25− T cells following in vitro exposure to antigen and IL-10, and the induced Treg cells themselves appear to mediate their inhibitory properties through the production of IL-10 and TGF-β. Tregs have been found to be fundamental for the control of autoimmune responses in several murine models such as inflammatory bowel disease, and depletion of CD25+ T cells has been shown to mediate immune rejection of various murine tumors in vivo
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


