(See also Chap. 148) In addition to receiving antibiotic prophylaxis, transplant recipients should be vaccinated against likely pathogens (Table 169-6). In the case of HSC transplant recipients, optimal responses cannot be achieved until after immune reconstitution, despite previous immunization of both donor and recipient. Recipients of an allogeneic HSC transplant must be reimmunized if they are to be protected against pathogens. The situation is less clear-cut in the case of autologous transplantation. T and B cells in the peripheral blood may reconstitute the immune response if they are transferred in adequate numbers. However, cancer patients (particularly those with Hodgkin’s disease, in whom vaccination has been extensively studied) who are undergoing chemotherapy do not respond normally to immunization, and titers of antibodies to infectious agents fall more rapidly than in healthy individuals. Therefore, even immunosuppressed patients who have not undergone HSC transplantation may need booster vaccine injections. If memory cells are specifically eliminated as part of a stem cell “cleanup” procedure, it will be necessary to reimmunize the recipient with a new primary series. Optimal times for immunizations of different transplant populations are being evaluated. Yearly immunization of household and other contacts (including health care personnel) against influenza benefits the patient by preventing local spread.
VACCINATION OF HEMATOPOIETIC STEM CELL TRANSPLANT (HSCT) AND SOLID ORGAN TRANSPLANT (SOT) RECIPIENTS |
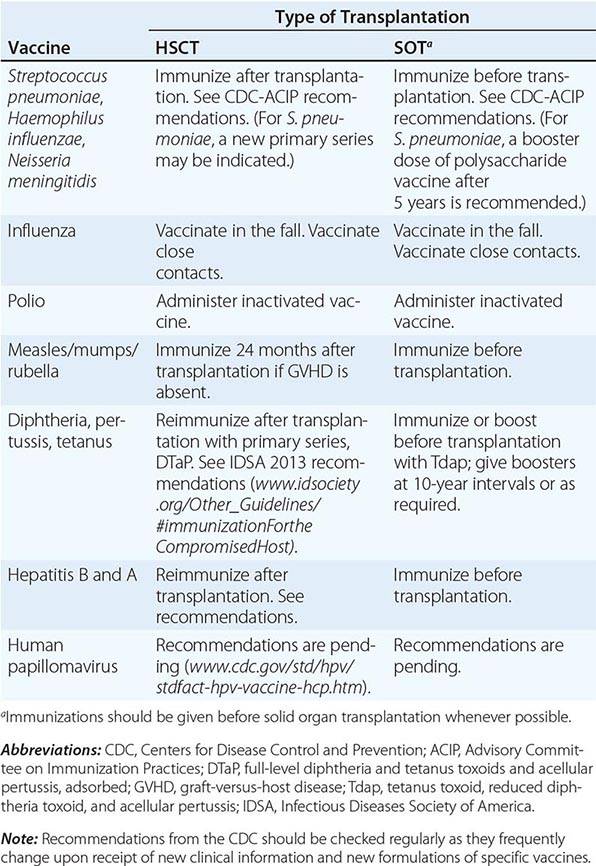
In the absence of compelling data as to optimal timing, it is reasonable to administer the pneumococcal and H. influenzae type b conjugate vaccines to both autologous and allogeneic HSC transplant recipients beginning 12 months after transplantation. A series that includes both the 13-valent pneumococcal conjugate vaccine (Prevnar) and the 23-valent pneumococcal polysaccharide vaccine (Pneumovax) is now recommended (according to CDC guidelines). The pneumococcal and H. influenzae type b vaccines are particularly important for patients who have undergone splenectomy. The Neisseria meningitidis polysaccharide conjugate vaccine (Menactra or Menveo) also is recommended. In addition, diphtheria, tetanus, acellular pertussis, and inactivated polio vaccines can all be given at these same intervals (12 months and, as required, 24 months after transplantation). Some authorities recommend a new primary series for tetanus/diphtheria/pertussis and inactivated poliovirus vaccines beginning 12 months after transplantation. Vaccination to prevent hepatitis B and hepatitis A (both killed vaccines) also seems advisable. A formal recommendation regarding immunization with the tetravalent HPV virus-like particle vaccine (Gardasil) after HSC transplantation has not been issued. However, HPV vaccination, which can prevent genital warts as well as specific cancers, is recommended through age 26 for healthy young adults who previously have not been vaccinated or have not received the full series. Live-virus measles/mumps/rubella (MMR) vaccine can be given to autologous HSC transplant recipients 24 months after transplantation and to most allogeneic HSC transplant recipients at the same point if they are not receiving maintenance therapy with immunosuppressive drugs and do not have ongoing GVHD. The risk of spread from a household contact is low for MMR vaccine. In parts of the world where live poliovirus vaccine is used, patients as well as contacts should be advised to receive only the killed vaccine. In the rare setting where both donor and recipient are VZV naïve and the recipient is no longer receiving acyclovir or ganciclovir prophylaxis, the patient should be counseled to receive varicella-zoster immune globulin (VariZIG) up to 10 days after an exposure to a person with chickenpox or uncovered zoster; such patients should avoid close contact with persons recently vaccinated with Varivax. A formal recommendation regarding Varivax immunization of such patients is not currently available. Neither patients nor their household contacts should receive vaccinia vaccine unless they have been exposed to smallpox virus. Among patients who have active GVHD and/or are taking high maintenance doses of glucocorticoids, it may be prudent to avoid all live-virus vaccines.
In the case of SOT recipients, administration of all the usual vaccines and of the indicated booster doses should be completed before immunosuppression, if possible, to maximize responses. For patients taking immunosuppressive agents, the administration of pneumococcal vaccine should be repeated every 5 years. No data are available for the meningococcal vaccine, but it is probably reasonable to administer it along with the pneumococcal vaccine. H. influenzae conjugate vaccine is safe and should be efficacious in this population; therefore, its administration before transplantation is recommended. Booster doses of this vaccine are not recommended for adults. SOT recipients who continue to receive immunosuppressive drugs should not receive live-virus vaccines. A person in this group who is exposed to measles should be given measles immune globulin. Similarly, an immunocompromised patient who is seronegative for varicella and who comes into contact with a person who has chickenpox should be given varicella-zoster immune globulin as soon as possible (optimally within 96 h; up to 10 days after contact); if this is not possible, a 10- to 14-day course of acyclovir therapy should be started immediately. Upon the discontinuation of treatment, clinical disease may still occur in a small number of patients; thus vigilance is indicated. Rapid re-treatment with acyclovir should limit the symptoms of disease. Household contacts of transplant recipients can receive live attenuated VZV vaccine, but vaccinees should avoid direct contact with the patient if a rash develops. Virus-like particle vaccines have been licensed for the prevention of infection with several HPV serotypes most commonly implicated in cervical and anal carcinomas and in anogenital and laryngeal warts. These vaccines are not live; however, no information is yet available about their immunogenicity or efficacy in transplant recipients.
Immunocompromised patients who travel may benefit from some but not all vaccines (Chaps. 148 and 149). In general, these patients should receive any killed or inactivated vaccine preparation appropriate to the area they are visiting; this recommendation includes the vaccines for Japanese encephalitis, hepatitis A and B, poliomyelitis, meningococcal infection, and typhoid. The live typhoid vaccines are not recommended for use in most immunocompromised patients, but an inactivated or purified polysaccharide typhoid vaccine can be used. Live yellow fever vaccine should not be administered. On the other hand, primary immunization or boosting with the purified-protein hepatitis B vaccine is indicated. Inactivated hepatitis A vaccine should also be used in the appropriate setting (Chap. 148). A vaccine is now available that provides dual protection against hepatitis A and hepatitis B. If hepatitis A vaccine is not administered, travelers should consider receiving passive protection with immune globulin (the dose depending on the duration of travel in the high-risk area).
SECTION 4 | APPROACH TO THERAPY FOR BACTERIAL DISEASES |
170 | Treatment and Prophylaxis of Bacterial Infections |
Antimicrobial agents have had a major impact on human health. Together with vaccines, they have contributed to reduced mortality, extended lifespan, and enhanced quality of life. Among drugs used in human medicine, however, they are distinctive in that their use promotes the occurrence of drug resistance in the pathogens they are designed to treat as well as in other “bystander” organisms. Indeed, the history of antimicrobial development has been driven in large part by the medical need engendered by the emergence of resistance to each generation of agents. Thus, the careful and appropriate use of antimicrobial drugs is particularly important not only for optimizing efficacy and minimizing adverse effects but also for minimizing the risk of resistance and preserving the value of existing agents. Although this chapter focuses on antibacterial agents, the optimal use of all antimicrobials depends on an understanding of each drug’s mechanism of action, spectrum of activity, mechanisms of resistance, pharmacology, and adverse effect profile. This information is then applied in the context of the patient’s clinical presentation, underlying conditions, and epidemiology to define the site and likely nature of the infection or other condition and thus to choose the best therapy. Gathering of microbiologic information is important for refining therapeutic choices on the basis of documented pathogen and susceptibility data whenever possible; this information also makes it possible to choose more targeted therapy, thereby reducing the risk of selection of resistant bacteria. Durations of therapy are chosen according to the nature of the infection and the patient’s response to treatment and are informed by clinical studies when they are available, with the understanding that shorter courses are less likely than longer ones to promote the emergence of resistance. This chapter provides specific information that is necessary for making informed choices among antibacterial agents.
MECHANISMS OF ACTION AND RESISTANCE
The mechanisms of action of and resistance to antibacterial agents are discussed in detail in the text and are summarized for the most commonly used groups of agents in Table 170-1. A schematic of antibacterial targets is provided in Fig. 170-1.
MECHANISMS OF ACTION OF AND RESISTANCE TO ANTIBACTERIAL AGENTS |
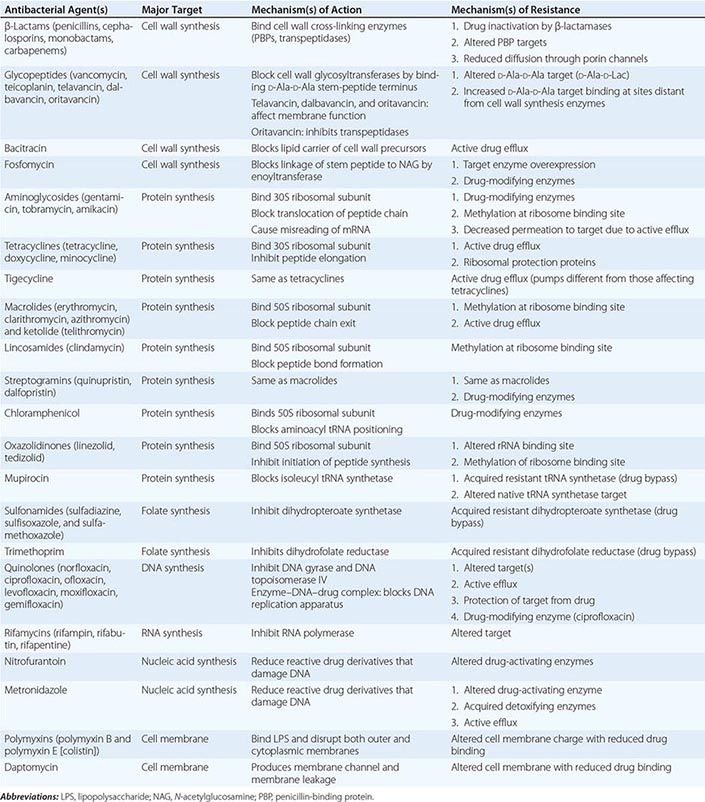
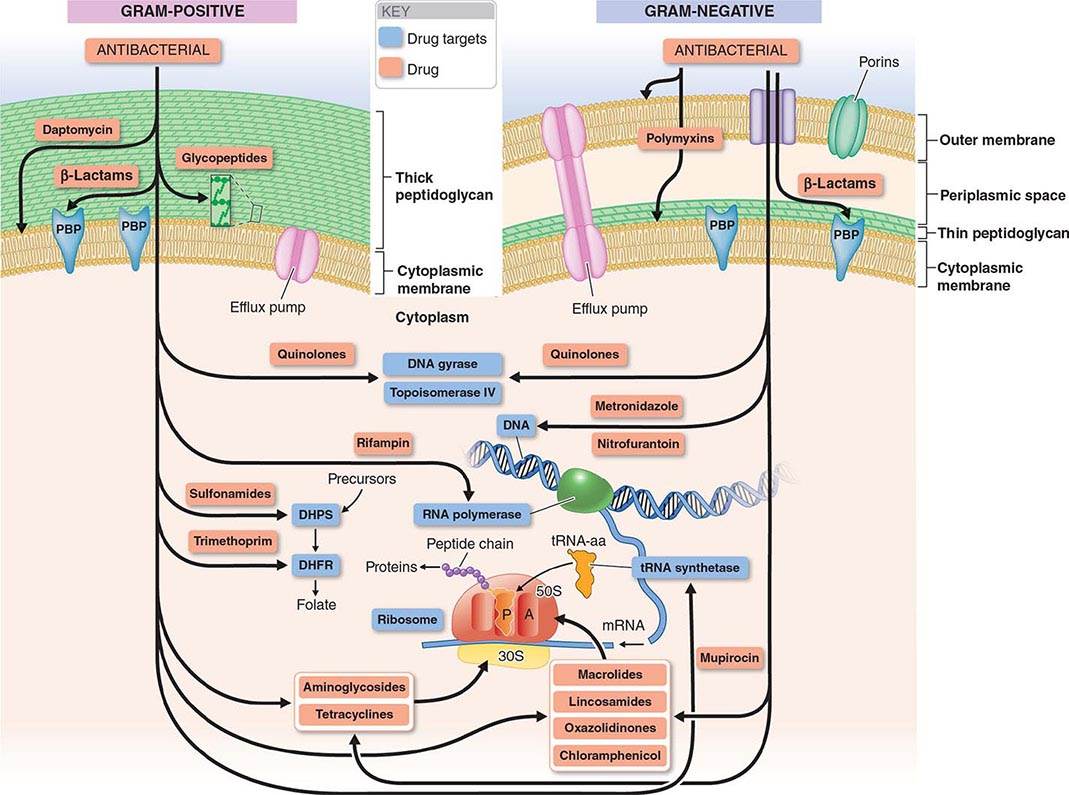
FIGURE 170-1 Antibacterial targets. A, aminoacyl site; DHFR, dihydrofolate reductase; DHPS, dihydropteroate synthetase; P, peptidyl site; PBP, penicillin-binding protein; tRNA-aa, aminoacyl tRNA.
MECHANISMS OF ACTION
Multiple essential components of bacterial cell structures and metabolism have been the targets of antibacterial agents used in clinical medicine, and the interaction of an agent with its target results in either inhibition of bacterial growth and replication (bacteriostatic effect) or bacterial killing (bactericidal effect). In general, targets have been chosen because they either do not exist in mammalian cells and physiology or are sufficiently different from their bacterial counterparts to allow selective bacterial targeting. Treatment with bacteriostatic agents is effective when the patient’s host defenses are sufficient to contribute to eradication of the infecting pathogen. In patients with impaired host defenses (e.g., neutropenia) or infections at body sites with impaired or limited host defenses (e.g., meningitis and endocarditis), bactericidal agents are generally preferred.
Inhibition of Cell Wall Synthesis The bacterial cell wall, which is external to the cytoplasmic membrane and has no counterpart in mammalian cells, protects bacterial cells from lysis under low osmotic conditions. The cell wall is a cross-linked peptidoglycan composed of a polymer of alternating units of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM), four-amino-acid stem peptides linked to each NAM, and a peptide cross-bridge that links adjacent stem peptides to form a netlike structure. Several steps in peptidoglycan synthesis are targets of antibacterial agents. Inhibition of cell wall synthesis generally results in a bactericidal effect that is linked to cell lysis. This effect results not only from the blocking of new cell-wall formation but from the uninhibited action of cell wall–remodeling enzymes called autolysins, which cleave peptidoglycan as part of normal cell-wall growth.
In gram-positive bacteria the peptidoglycan is the most external cell structure, but in gram-negative bacteria an asymmetric lipid outer membrane is external to the peptidoglycan and contains diffusion channels called porins. The space between the cytoplasmic membrane peptidoglycan and the outer membrane is referred to as the periplasmic space. Most antibacterial drugs enter the gram-negative bacterial cell through a porin channel, since the outer membrane is a major diffusion barrier. Although the peptidoglycan layer is thicker in gram-positive (20–80 nm) than in gram-negative (1 nm) bacteria, peptidoglycan itself constitutes only a limited diffusion barrier for antibacterial agents.
β-LACTAMS The β-lactam drugs, including penicillins, cephalosporins, monobactams, and carbapenems, target transpeptidase enzymes (also called penicillin-binding proteins, or PBPs) involved in the stem-peptide cross-linking step.
GLYCOPEPTIDES The glycopeptides, including vancomycin, teicoplanin, telavancin, dalbavancin, and oritavancin, bind the two terminal D-alanine residues of the stem peptide, hindering the glycosyltransferase involved in polymerizing NAG–NAM units. Telavancin also binds to the lipid II intermediate that delivers cell-wall precursor subunits. Likewise, dalbavancin and oritavancin interact with the cell membrane, and oritavancin may also inhibit transpeptidases. Both β-lactams and glycopeptides interact with their targets external to the cytoplasmic membrane.
BACITRACIN (TOPICAL) AND FOSFOMYCIN These agents interrupt enzymatic steps in the production of peptidoglycan precursors in the cytoplasm.
Inhibition of Protein Synthesis Most inhibitors of bacterial protein synthesis target bacterial ribosomes, whose difference from eukaryotic ribosomes allows selective antibacterial action. Some inhibitors bind to the 30S ribosomal subunit and others to the 50S subunit. Most protein synthesis–inhibiting agents are bacteriostatic; aminoglycosides are an exception and are bactericidal.
AMINOGLYCOSIDES Aminoglycosides (amikacin, gentamicin, kanamycin, netilmicin, streptomycin, tobramycin) bind irreversibly to 16S ribosomal RNA (rRNA) of the 30S ribosomal subunit, blocking the translocation of peptidyl transfer RNA (tRNA) from the A (aminoacyl) to the P (peptidyl) site and, at low concentrations, causing misreading of messenger RNA (mRNA) codons and thus causing the introduction of incorrect amino acids into the peptide chain; at higher concentrations, translocation of the peptide chain is blocked. Cellular uptake of aminoglycosides is dependent on the electrochemical gradient across the bacterial membrane. Under anaerobic conditions, this gradient is reduced, with a consequent reduction in the uptake and activity of the aminoglycosides. Spectinomycin is a related aminocyclitol antibiotic that also binds to 16S rRNA of the 30S ribosomal subunit but at a different site. This drug inhibits translocation of the growing peptide chain but does not trigger codon misreading and produces only a bacteriostatic effect.
TETRACYCLINES AND GLYCYLCYCLINES Tetracyclines (doxycycline, minocycline, tetracycline) bind reversibly to the 16S rRNA of the 30S ribosomal subunit and block the binding of aminoacyl tRNA to the ribosomal A site, thereby inhibiting peptide elongation. Active transport of tetracyclines into bacterial but not mammalian cells contributes to the selectivity of these agents. Tigecycline, a derivative of minocycline and the only available glycylcycline, acts similarly to the tetracyclines but is distinctive for its ability to circumvent the most common mechanisms of resistance to the tetracyclines.
MACROLIDES AND KETOLIDES In contrast to the aminoglycosides and tetracyclines, the macrolides (azithromycin, clarithromycin, erythromycin) and ketolides (telithromycin) bind to the 23S rRNA of the 50S ribosomal subunit. These agents block translocation of the growing peptide chain by binding to the tunnel from which the chain exits the ribosome.
LINCOSAMIDES Clindamycin is the only lincosamide in clinical use. It binds to the 23S rRNA of the 50S ribosomal subunit, interacting with both the ribosomal A and P sites and blocking peptide bond formation.
STREPTOGRAMINS The only streptogramin in clinical use is a combination of quinupristin, a group B streptogramin, and dalfopristin, a group A streptogramin. Both components bind to 23S rRNA of the 50S ribosome: dalfopristin binds to both the A and P sites of the peptidyl transferase center, and quinupristin binds to a site that overlaps the macrolide-binding site, blocking the emergence of nascent peptide from the ribosome. The combination is bactericidal, but macrolide-resistant bacteria exhibit cross-resistance to quinupristin, and the remaining activity of dalfopristin alone is bacteriostatic.
CHLORAMPHENICOL Chloramphenicol binds reversibly to the 23S rRNA of the 50S subunit in a manner that interferes with the proper positioning of the aminoacyl component of tRNA in the A site. This site of binding is near those of the macrolides and lincosamides.
OXAZOLIDINONES Linezolid and tedizolid are the only oxazolidinones in clinical use. They bind directly to the A site in the 23S rRNA of the 50S ribosomal subunit and block binding of aminoacyl tRNA, inhibiting the initiation of protein synthesis.
MUPIROCIN Mupirocin (pseudomonic acid) is used topically. It competes with isoleucine for binding to isoleucyl tRNA synthetase, depleting stores of isoleucyl tRNA and thereby inhibiting protein synthesis.
Inhibition of Bacterial Metabolism Available inhibitors (antimetabolites) target the pathway for synthesis of folate, which is a cofactor in a number of one-carbon transfer reactions involved in the synthesis of some nucleic acids, including pyrimidine, thymidine, and all purines (adenine and guanine), as well as some amino acids (methionine and serine) and acetyl coenzyme A (CoA). Two sequential steps in folate synthesis are targeted. The selective antibacterial effect stems from the inability of mammalian cells to synthesize folate; they depend instead on exogenous sources. Antibacterial activity, however, may be reduced in the presence of high exogenous concentrations of the end products of the folate pathway (e.g., thymidine and purines) that may occur in some infections, resulting from local breakdown of leukocytes and host tissues.
SULFONAMIDES Sulfonamides, including sulfadiazine, sulfisoxazole, and sulfamethoxazole, inhibit dihydropteroate synthetase, which adds p-aminobenzoic acid (PABA) to pteridine, producing dihydropteroate. Sulfonamides are structural analogues of PABA and act as competing enzyme substrates.
TRIMETHOPRIM Subsequent steps in folate synthesis are catalyzed by dihydrofolate synthase, which adds glutamate to dihydropteroate, and dihydrofolate reductase, which then generates the final product, tetrahydrofolate. Trimethoprim is a structural analogue of pteridine and inhibits dihydrofolate reductase. Trimethoprim is available alone but is most often used in combination products that also contain sulfamethoxazole and thus block two sequential steps in folate synthesis.
Inhibition of DNA and RNA Synthesis or Activity A variety of antibacterial agents act on these processes.
QUINOLONES The quinolones include nalidixic acid, the first agent in the class, and newer, more widely used fluorinated derivatives (fluoroquinolones), including norfloxacin, ciprofloxacin, levofloxacin, moxifloxacin, and gemifloxacin. The quinolones are synthetic compounds that inhibit bacterial DNA synthesis by interacting with the DNA complexes of two essential enzymes, DNA gyrase and DNA topoisomerase IV, which alter DNA topology. Quinolones trap enzyme–DNA complexes in such a way that they block movement of the DNA replication apparatus and can generate lethal double-strand breaks in DNA, resulting in bactericidal activity. Although mammalian cells also have type II DNA topoisomerases like gyrase and topoisomerase IV, the structures of the mammalian enzymes are sufficiently different from those of the bacterial enzymes that quinolones have substantially selective antibacterial activity.
RIFAMYCINS Rifampin, rifabutin, and rifapentine are semisynthetic derivatives of rifamycin B and bind the β subunit of bacterial RNA polymerase, thereby blocking elongation of mRNA. Their action is highly selective for the bacterial enzyme over mammalian RNA polymerases.
NITROFURANTOIN The reduction of nitrofurantoin, a nitrofuran compound, by bacterial enzymes produces highly reactive derivatives that are thought to cause DNA strand breakage. Nitrofurantoin is used only for the treatment of lower urinary tract infections.
METRONIDAZOLE Metronidazole is a synthetic nitroimidazole with activity limited to anaerobic bacteria and certain anaerobic protozoa. Reduction of its nitro group by the electron-transport system in anaerobic bacteria produces reactive intermediates that damage DNA and result in bactericidal activity. Both nitrofurantoin and metronidazole have selective antibacterial activity because the reducing activity needed to generate active derivatives is generated only by bacterial and not mammalian enzymes.
Disruption of Membrane Integrity The integrity of the bacterial cytoplasmic membrane—and, in gram-negative bacteria, the outer membrane—is important for bacterial viability. Two bactericidal drugs have membrane targets.
POLYMYXINS The polymyxins, including polymyxin B and polymyxin E (colistin), are cationic cyclic polypeptides that disrupt the cytoplasmic membrane and the outer membrane (the latter by binding lipopolysaccharide).
DAPTOMYCIN Daptomycin is a lipopeptide that binds the cytoplasmic membrane of gram-positive bacteria in the presence of calcium, generating a channel that leads to leakage of cytoplasmic potassium ions and membrane depolarization.
MECHANISMS OF RESISTANCE
Bacteria use a wide variety of mechanisms to block or circumvent the activity of antibacterial agents. Although myriad, these mechanisms can generally be grouped into three categories: (1) altered or bypass targets that exhibit reduced binding of the drug, (2) altered access of the drug to its target by reductions in uptake or increases in active efflux, and (3) a modification of the drug that reduces its activity. These mechanisms result from either mutations in bacterial chromosomal genes occurring spontaneously during bacterial DNA replication or the acquisition of new genes by DNA transfer from other bacteria or uptake of exogenous DNA. New genes are most often acquired on self-replicating plasmids or other DNA elements transferred from other bacteria. However, some bacteria, such as Streptococcus pneumoniae and Neisseria gonorrhoeae, can also take up fragments of environmental DNA from related species and recombine that DNA directly into their own chromosomes, a process called transformation. Not uncommonly, resistant bacteria have combinations of resistance mechanisms either within one category or among categories, and many plasmids contain more than one resistance gene. Thus, plasmid acquisition itself can in many cases confer resistance to multiple antibacterial agents.
Many antibacterial drugs are derived from natural products of microbial species. Some genes encoding resistance to these drugs may have evolved and been mobilized onto plasmids from a protection mechanism in the producer organism or in other surviving bacteria in the exposed environment. Exposure to antibacterial agents either in nature or from human or animal use results in the selection of resistant strains within an otherwise susceptible bacterial population. Because the patterns and extent of resistance may differ among settings, initial choices of antibacterial drugs should be based, whenever possible, on local susceptibility data and should be modified as needed as soon as specific microbiology susceptibility data become available.
β-Lactams The most common mechanism of resistance to β-lactams is their degradation by β-lactamases, enzymes that break down the core β-lactam ring and destroy drug activity. Different β-lactamases degrade different β-lactams. Some β-lactamases are encoded on the bacterial chromosome, and their activity contributes to the susceptibility profile of a particular species. Because other β-lactamases are encoded by acquired plasmids, their resistance profiles may be present in some strains of a species but not others. In gram-positive bacteria β-lactamases are secreted into the extracellular environment, whereas in gram-negative bacteria these enzymes are secreted into the periplasmic space between the cytoplasmic and outer membranes. Thus, in gram-negative bacteria, access of β-lactams both to their target PBPs and to β-lactamases requires diffusion across the outer membrane, generally through the porin channels.
Most strains of Staphylococcus aureus produce a plasmid-encoded β-lactamase that degrades penicillin but not semisynthetic penicillins, such as oxacillin and nafcillin. The most common plasmid-encoded β-lactamases of gram-negative bacteria are able to inactivate all penicillins and most earlier-generation cephalosporins. Extended-spectrum β-lactamase (ESBL) variants of these early enzymes that can degrade later-generation cephalosporins (ceftriaxone, cefotaxime, ceftazidime) as well as the monobactam aztreonam have now emerged and are widely disseminated. Some ESBLs also degrade the fourth-generation cephalosporin cefepime. Carbapenems (imipenem, meropenem, ertapenem, doripenem) are not degraded by ESBLs, but additional β-lactamases, called carbapenemases, that degrade carbapenems and most if not all other β-lactams have begun to emerge.
The chromosomal β-lactamase of Klebsiella pneumoniae preferentially degrades penicillins but not cephalosporins. In contrast, the chromosomal β-lactamase of Enterobacter and related genera, AmpC, can degrade almost all cephalosporins but is normally expressed in small amounts. Mutations that cause increased amounts of AmpC to be produced confer full resistance to penicillins and cephalosporins; the exceptions are cefoxitin and cefepime, which are relatively stable to AmpC. Resistance to cefepime can develop, however, through the combined effects of increased AmpC production and decreased porin diffusion channels. Genes encoding AmpC have also been found on plasmids but are less common than plasmid-encoded ESBLs.
Inhibitors of β-lactamases such as clavulanate, sulbactam, and tazobactam have been developed and paired with amoxicillin and ticarcillin, ampicillin, and piperacillin, respectively. These inhibitors have little or no antibacterial activity of their own but inhibit plasmid-mediated β-lactamases, including ESBLs but not AmpC enzymes.
Resistance to β-lactams also occurs through alterations in their target PBPs. In S. pneumoniae, N. gonorrhoeae, and Neisseria meningitidis, resistance to penicillin occurs by recombination of transformed DNA from related species. In staphylococci, resistance to methicillin and other β-lactams occurs by the acquisition of the mec gene, which encodes PBP2a with reduced drug affinity. Ceftaroline is the only β-lactam that has affinity for PBP2a and is thus active against methicillin-resistant staphylococcal strains.
Glycopeptides Resistance to vancomycin in enterococci is due to the acquisition of a set of van genes that result in (1) the production of D-alanine-D-lactate—instead of the normal D-alanine-D-alanine—at the end of the peptidoglycan stem peptide and (2) the reduction of existing D-alanine-D-alanine terminated peptides. Vancomycin binds D-alanine-D-lactate with a thousandfold lower affinity than D-alanine-D-alanine. In a small number of cases, the van gene cassettes have been transferred from enterococci to S. aureus, with the consequent generation of full vancomycin resistance. Particularly in patients receiving prolonged courses of vancomycin, intermediate resistance to this drug has developed in S. aureus by a different mechanism: multiple chromosomal mutations that result in a thickened and poorly cross-linked cell wall in which multiple distant D-alanine-D-alanine stem peptide termini exist and bind vancomycin, impeding its access to the binding sites proximal to the cell membrane where new cell-wall synthesis occurs and where binding would block transpeptidase and transglycosylase enzymes. Susceptibility to telavancin, dalbavancin, and oritavancin is also reduced in strains that exhibit resistance or intermediate susceptibility to vancomycin, although in some cases strains may still be classified as susceptible on the basis of clinical interpretive criteria.
Aminoglycosides The most common mechanism of resistance is due to acquisition of plasmid genes encoding transferase enzymes that modify aminoglycosides by the addition of acetyl, adenyl, or phosphate groups; these added groups decrease the drugs’ binding affinity to their ribosomal target site. Transferases differ in which aminoglycosides they modify, and amikacin resistance occurs less often than resistance to gentamicin or tobramycin. More recently, plasmids encoding methyltransferases that modify the ribosomal site of aminoglycoside binding and confer resistance to all aminoglycosides have been found in enteric gram-negative bacteria. For streptomycin, a ribosomal protein mutation may cause resistance. In Pseudomonas aeruginosa, resistance may also occur through mutations causing increased expression of a chromosomally encoded efflux pump, MexXY.
Tetracyclines and Glycylcyclines For tetracyclines, resistance is most often plasmid mediated and attributable either to active efflux pumps, which are generally specific for tetracyclines, or to proteins that protect the ribosome from tetracycline action. Resistance to the glycylcycline tigecycline, which is not affected by the usual tetracycline resistance mechanisms, can occur through mutations that cause overexpression of certain broad-spectrum efflux pumps in Proteus species.
Macrolides, Ketolides, Lincosamides, and Streptogramins Resistance to macrolides, clindamycin, and quinupristin is most often due to plasmid-acquired methylases that modify the drug binding site on the ribosome. Resistance to quinupristin by this mechanism renders the quinupristin-dalfopristin combination bacteriostatic rather than bactericidal. Telithromycin, a ketolide, has an additional binding site on the ribosome and remains active in the presence of these methylases. Acquired genes encoding active efflux pumps can also contribute to resistance to macrolides in streptococci and resistance to macrolides, clindamycin, and dalfopristin in staphylococci. Plasmid-acquired drug-modifying enzymes in staphylococci can also cause resistance to quinupristin and dalfopristin. Macrolide resistance due to 23S rRNA mutations is uncommon in staphylococci and streptococci because of the multiple copies of the rRNA genes on the chromosomes of these species; such resistance may occur more frequently, however, in mycobacteria and Helicobacter pylori, which have only single chromosomal copies of these rRNA genes.
Chloramphenicol Resistance to chloramphenicol is most often due to a plasmid-encoded drug-modifying acetyltransferase.
Oxazolidinones Linezolid resistance has been seen in enterococci more often than in staphylococci and, in both organisms, is due to mutations in multiple copies of the 23S rRNA genes that reduce drug binding to the ribosome. A plasmid-acquired ribosomal methylase gene that confers resistance to both chloramphenicol and linezolid has also been found in some strains of staphylococci but is not yet widespread. Tedizolid may still be active against some but not all linezolid-resistant strains.
Mupirocin Resistance to mupirocin occurs by either mutation in the target leucyl-tRNA synthetase (low-level resistance) or the acquisition of a plasmid-encoded resistant tRNA synthetase (high-level resistance).
Sulfonamides and Trimethoprim Resistance to both of these antimetabolites is due to plasmid-acquired genes encoding resistant enzymes that bypass the inhibition of the native sensitive enzymes—a resistant dihydropteroate synthetase in the case of sulfonamides and a resistant dihydrofolate reductase in the case of trimethoprim.
Quinolones Resistance to quinolones is most often due either to chromosomal mutations altering the target enzymes DNA gyrase and DNA topoisomerase IV, with consequent reduction in drug binding, or to mutations that increase the expression of native broad-spectrum efflux pumps for which quinolones (among other compounds) are substrates. In addition, three types of genes can confer reduced susceptibility or low-level resistance by protecting target enzymes, modifying some quinolones, or pumping quinolones out of the cell (efflux). These genes are located on multidrug resistance plasmids that have spread worldwide. Their presence can promote the selection of higher levels of quinolone resistance linked to resistance to other antibacterial drugs that is encoded on the same plasmid.
Rifampin and Rifabutin Single mutations in the β subunit of RNA polymerase can cause high-level resistance to rifampin. Thus rifampin and other rifamycins are used for treatment of infections only in combination with other antibacterial drugs in order to prevent resistance.
Metronidazole Acquired resistance to metronidazole in Bacteroides species is rare. Such resistance has been reported in strains that lack endogenous nitroreductase activity or that have acquired nim genes responsible for further reduction of DNA-damaging nitroso intermediates to an inactive derivative. Active efflux and enhanced DNA repair mechanisms also have been associated with resistance.
Nitrofurantoin Resistance to nitrofurantoin in Escherichia coli can emerge through a series of mutations that progressively decrease the nitroreductase activity necessary for generating active nitrofuran metabolites.
Polymyxins Because of emerging multidrug resistance in gram-negative bacteria, colistin and polymyxin B are being used increasingly for infections due to resistant Enterobacteriaceae, P. aeruginosa, and Acinetobacter species. Rates of resistance vary. Resistance can emerge during therapy through mutations that cause reductions in the negative charge of the gram-negative bacterial cell surface, thereby reducing binding of the positively charged colistin.
Daptomycin The mechanisms of resistance to daptomycin are complex and involve mutations in several genes that can alter cell membrane charge and reduce daptomycin binding. Resistance to daptomycin is relatively infrequent but has emerged in some S. aureus strains with intermediate vancomycin susceptibility from patients treated with vancomycin but not with daptomycin. In some methicillin-resistant S. aureus (MRSA) strains, daptomycin resistance has been linked to acquired susceptibility to β-lactams; combinations of daptomycin and nafcillin have been successful for treatment of patients infected with resistant strains when daptomycin alone or in combination with other agents has failed. The mechanism of this effect is not yet clear.
PHARMACOKINETICS AND PHARMACODYNAMICS
The term pharmacokinetics describes the disposition of a drug in the body, whereas pharmacodynamics describes the determinants of drug action on the pathogen in relation to pharmacokinetic factors. An understanding of the principles governing these two areas is required for effective drug selection, dosing, and prevention of toxicities.
PHARMACOKINETICS
The process of drug disposition has four principal phases: absorption, distribution, metabolism, and excretion. These components determine the time course of drug concentrations in serum and subsequently the concentrations in other tissues and body fluids.
Absorption When a drug is given by a particular route, absorption is defined as the percentage of the dose that reaches the systemic circulation. For example, since IV administration provides direct access to the systemic circulation, 100% of a drug dose given IV is usually absorbed. The level of absorption becomes more relevant when non-IV routes are used—e.g., the oral, IM, SC, and topical routes. The percentage of a drug that is absorbed is termed its bioavailability. Examples of antibacterial agents with a high oral bioavailability include metronidazole, levofloxacin, and linezolid. IV administration and oral dosing for highly bioavailable agents usually give equivalent results. Many factors can affect a drug’s oral bioavailability, including the timing of food consumption relative to drug administration, drug-metabolizing enzymes, efflux transporters, concentration-dependent solubility, and acid degradation. Underlying conditions such as diarrhea or ileus can also affect the site of drug absorption and thereby alter bioavailability. Certain orally administered drugs have lower bioavailability because of the first-pass effect—the process by which drugs are absorbed in the small intestine through the portal circulation and then directly transported to the liver for metabolism.
Distribution Distribution describes the process by which a drug transfers reversibly between the general circulation and the tissues. After absorption into the general circulation and the central compartment (the extensively perfused organs), the drug will also distribute into the peripheral compartment (less well-perfused tissues). The volume of distribution (Vd) is a pharmacokinetic parameter that describes the amount of drug in the body at a given time relative to the measured serum concentration. Properties such as the drug’s lipophilicity, partition coefficient within different body tissues, and protein binding; blood flow; and pH can affect the volume of distribution. Drugs with a small volume of distribution are limited to certain areas within the body (typically extracellular fluid), whereas those with a higher volume of distribution penetrate extensively into tissues throughout the body. Antibacterial drugs can bind to serum proteins, and a given drug is usually described as either poorly or highly protein bound. Only the unbound (free) drug is active and available to exert antibacterial effects. For example, because tigecycline is highly protein bound and also has a large volume of distribution, concentrations of free drug in the serum are low.
Metabolism Metabolism is the chemical transformation of a drug by the body. This modification can occur within several areas; the liver is the organ most commonly involved. Drugs are metabolized by enzymes, but enzyme systems have a finite capacity to metabolize a substrate drug. If a drug is given in a dose at which the concentration does not exceed the rate of metabolism, then the metabolic process is generally linear. If the dose exceeds the amount that can be metabolized, drug accumulation and potential toxicity may occur. Drugs are metabolized through phase I or phase II reactions. In phase I reactions, the drug is made more polar through dealkylation, hydroxylation, oxidation, and deamination. Polarity facilitates drug removal from the body. Phase II reactions, which include glucuronidation, sulfation, and acetylation, result in compounds larger and more polar than the parent drug. Both phases usually inactivate the parent drug, but some drugs are rendered more active. The cytochrome P450 (CYP) enzyme system is responsible for phase I reactions and is generally found in the liver. CYP3A4 is a common subfamily within this system that is responsible for the majority of drug metabolism. Antibacterial drugs can be substrates, inhibitors, or inducers of a particular CYP enzyme. Inducers, such as rifampin, can increase the production of CYP enzymes and consequently increase the metabolism of other drugs. Inhibitors, such as quinupristin-dalfopristin, cause a decrease in enzyme activity (or competition for CYP substrate) and therefore an increase in the concentration of the interacting drug.
Excretion Excretion describes the body’s mechanisms of drug elimination. Drugs can be eliminated through more than one mechanism. Renal clearance is the most common route and includes elimination through glomerular filtration, tubular secretion, and/or passive diffusion. Some agents have nonrenal clearance and rely on the biliary tree or the intestine for excretion. Excretion affects the half-life of a drug—i.e., the time it takes for the blood concentration of a drug to decrease by one-half. This value can range from minutes to days. Approximately five to seven half-lives are required for a drug to reach steady state when multiple doses are given in a time frame shorter than the half-life itself. Drug half-life and overall clearance can be extended if the organ responsible for clearance is impaired. Patients with renal or hepatic impairment may require dose adjustments that take delayed clearance into account and prevent toxicities from drug accumulation. For example, imipenem is cleared predominantly through glomerular filtration, and in the presence of renal impairment the dosing interval is typically increased to account for the increased half-life.
PHARMACODYNAMICS
The term pharmacodynamics describes the relationship between the serum concentrations that determine the efficacy of the drug and the serum concentrations that produce the toxic effects of the drug. For an antibacterial agent, the pharmacodynamic focus is the type of drug exposure needed for optimal antibacterial effect in relation to the minimal inhibitory concentration (MIC)—the lowest drug concentration that inhibits the visible growth of a microorganism under standardized laboratory conditions. Antibacterial effect usually correlates with one of the following parameters: (1) ratio of peak serum concentration to the MIC (Cmax/MIC), (2) ratio of the area under the concentration–time curve to the MIC (AUC/MIC), or (3) duration of concentrations above the MIC (T>MIC) (Fig. 170-2).
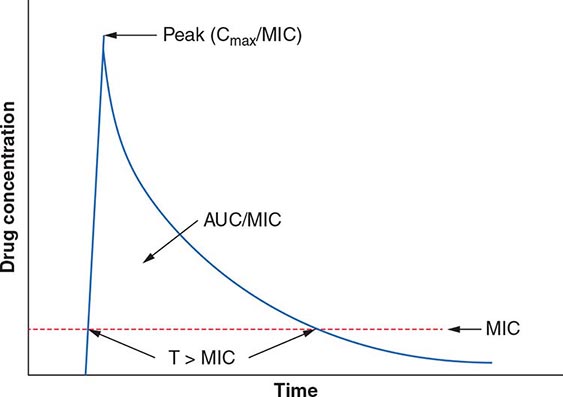
FIGURE 170-2 Pharmacokinetic and pharmacodynamic model predicting efficacy of antibacterial drugs. AUC, area under the time–concentration curve; Cmax, peak serum concentration of drug; MIC, minimal inhibitory concentration; T>MIC, duration of drug concentrations above the MIC.
For concentration-dependent killing agents, as the designation implies, the higher the drug concentration, the higher the rate and extent of bacterial killing. Aminoglycosides fit into the Cmax/MIC model of pharmacodynamics activity, and a particular peak serum concentration is often targeted to achieve optimal killing. Fluoroquinolones exemplify antibacterial agents for which the AUC/MIC is a predictor of efficacy. For example, studies have found that an AUC/MIC ratio of >30 will maximize killing of S. pneumoniae by fluoroquinolones. In contrast, time-dependent killing agents reach a ceiling at which higher concentrations do not result in increased effect. Instead, these agents are active against bacteria only when the drug concentration is above the MIC. The T>MIC predicts clinical efficacy for all β-lactams. The longer the concentration of the β-lactam remains above the MIC for an infecting pathogen during the dosing interval, the greater the killing effect. For some drug classes, such as aminoglycosides, a postantibiotic effect—the delayed regrowth of surviving bacteria after exposure to an antibiotic—supports less frequent dosing.
APPROACH TO THERAPY
The approach to antibiotic therapy is driven by host factors, site of infection, and local resistance profiles of suspected or known pathogens. Further, national and local drug shortages and formulary restrictions can affect available therapies. Regular monitoring of the patient and collection of laboratory data should be undertaken to streamline antibacterial therapy as appropriate and to investigate the possibility of treatment failure if the patient fails to respond appropriately.
EMPIRICAL AND DIRECTED THERAPY
Therapy is considered empirical when the causative agent has yet to be determined and therapeutic decisions are based on the severity of illness and the clinician’s assessment of likely pathogens in light of the clinical syndrome, the patient’s medical conditions and prior therapy, and relevant epidemiologic factors. For patients with severe illness, empirical therapy often takes the form of an antibacterial combination that provides broad coverage of diverse agents and thus ensures adequate treatment of possible pathogens while additional data are being collected. Directed therapy is predicated on identification of the pathogen, determination of its susceptibility profile, and establishment of the extent of the infection. Directed therapy generally allows the use of more targeted and narrower-spectrum antibacterial agents than does empirical therapy.
Information on epidemiology, exposures, and local antibacterial susceptibility patterns can help guide empirical therapy. When empirical treatment is clinically appropriate, care should be taken to obtain clinical specimens for microbiologic analysis before the initiation of therapy and to de-escalate therapy as new information is obtained about the patient’s clinical condition and the causal pathogens. De-escalation to the point of directed therapy can limit unnecessary risks to the patient as well as the risk of emergence of antibacterial resistance.
SITE OF INFECTION
The site of infection is a consideration in antibacterial therapy, largely because of the differing abilities of drugs to penetrate and achieve adequate concentrations at particular body sites. For example, to be effective in the treatment of meningitis, an agent must (1) be able to cross the blood–brain barrier and reach adequate concentrations in the cerebrospinal fluid (CSF) and (2) be active against the relevant pathogen(s). Dexamethasone, administered with or 15–20 min before the first dose of an antibacterial drug, has been shown to improve outcomes in patients with acute bacterial meningitis, but its use may reduce penetration of some antibacterial agents, such as vancomycin, into the CSF. In this case, rifampin is added because its penetration is not reduced by dexamethasone. Infections at other sites where either pathogens are protected from normal host defenses or penetration of an antibacterial drug is suboptimal include osteomyelitis, prostatitis, intraocular infections, and abscesses. In such cases, consideration must be given to the mechanism of drug delivery (e.g., intravitreal injections) as well as to the role of interventions to drain, debride, or otherwise reduce the barriers to effective antibacterial therapy.
HOST FACTORS
Host factors, including immune function, pregnancy, allergies, age, renal and hepatic function, drug–drug interactions, comorbid conditions, and occupational or social exposures, should be considered.
Immune Dysfunction Patients with deficits in immune function that blunt the response to bacterial infection, including neutropenia, deficient humoral immunity, and asplenia (either surgical or functional), are all at increased risk of severe bacterial infection. Such patients should be treated aggressively and often broadly in the early stages of suspected infection pending results of microbiologic tests. For asplenic patients, treatment should include coverage of encapsulated organisms, particularly S. pneumoniae, that may cause rapidly life-threatening infection.
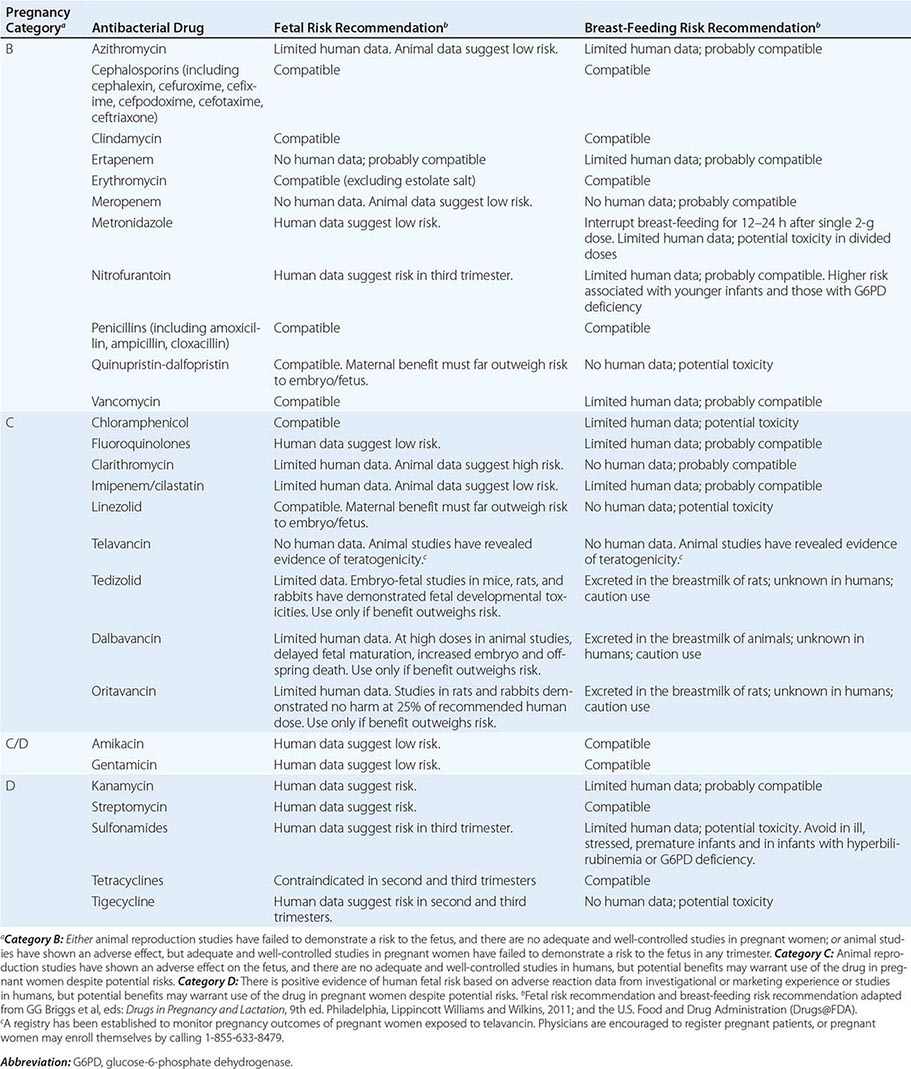
Pregnancy Pregnancy affects decisions regarding antibacterial therapy in two respects. First, pregnancy is associated with an increased risk of particular infections (e.g., those caused by Listeria). Second, the potential risks to the fetus that are posed by specific drugs must be considered. As for other drugs, the safety of the vast majority of antibacterial agents in pregnancy has not been established, and such agents are grouped in categories B and C by the U.S. Food and Drug Administration. Drugs in categories D and × are contraindicated in pregnancy or lactation due to established risks. The risks associated with antibacterial use in pregnancy and during lactation are summarized in Table 170-2.
RISKS ASSOCIATED WITH USE OF ANTIBACTERIAL DRUGS IN PREGNANCY AND LACTATION |
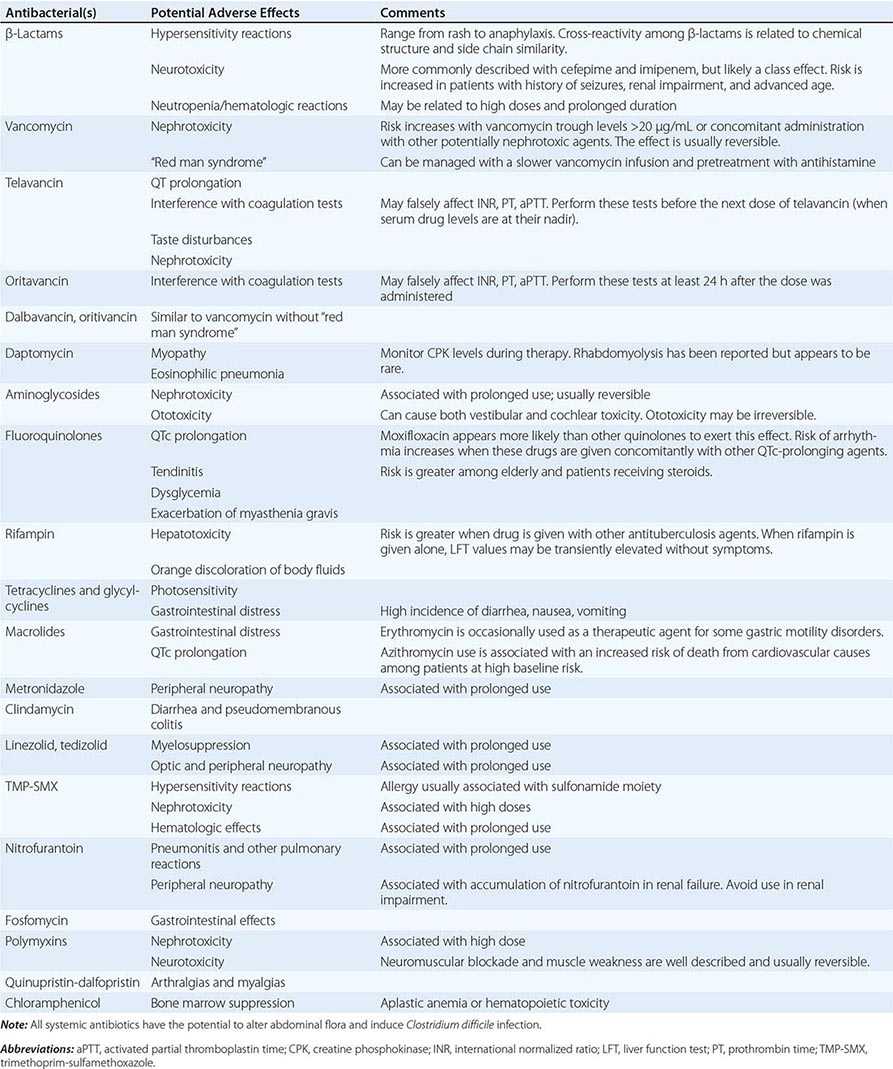
Allergies Allergies to antibiotics are among the most common allergies reported, and an allergy history should be obtained whenever possible before therapy is chosen. A detailed allergy history can shed light on the type of reaction experienced previously and on whether rechallenge with the same or a related medication is advisable (and, if so, under what circumstances). Allergies to the penicillins are most common. Although as many as 10% of patients may report an allergy to penicillin, studies suggest that up to 90% of these patients could tolerate a penicillin or cephalosporin. Adverse effects (Table 170-3) should be distinguished from true allergies to ensure appropriate selection of antibacterial therapy.
COMMON ADVERSE REACTIONS TO ANTIBACTERIAL AGENTS |
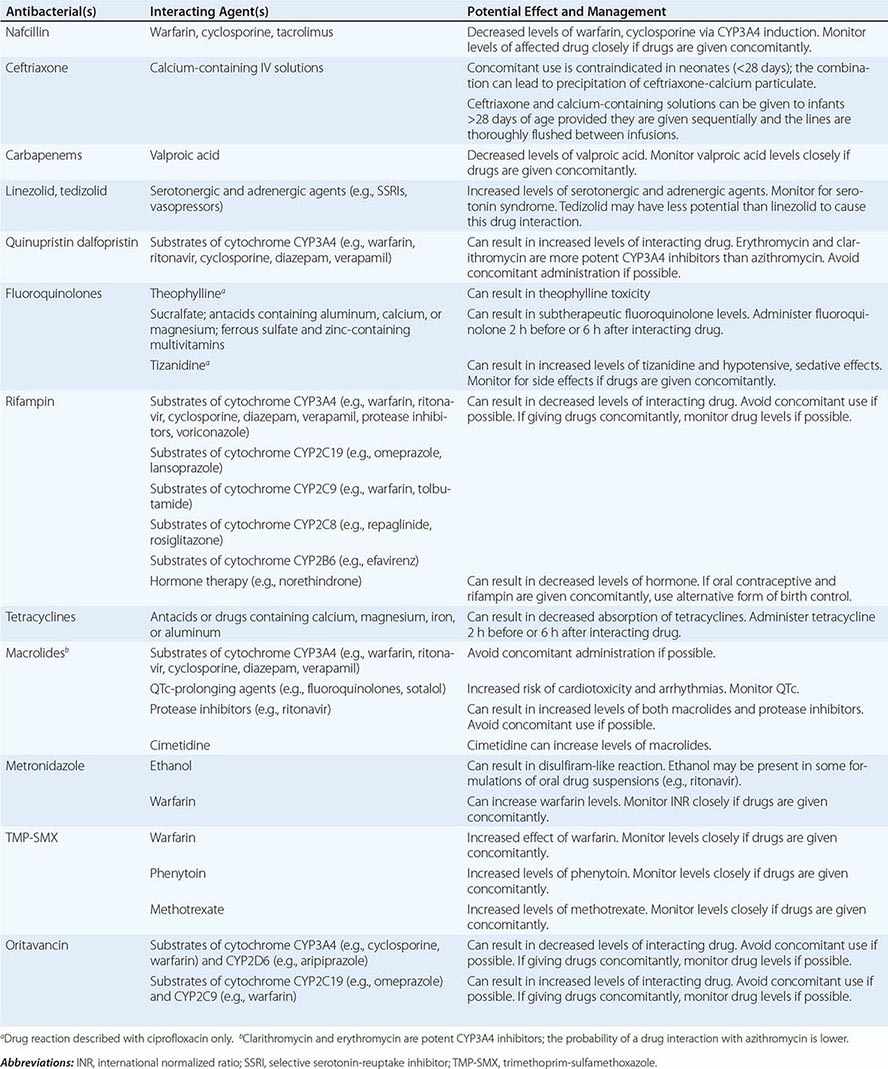
Drug–Drug Interactions Patients commonly receive other drugs that may interact with antibacterial agents. A summary of the most common drug–drug interactions, by antibacterial class, is provided in Table 170-4.
SIGNIFICANT ANTIBACTERIAL DRUG INTERACTIONS |
Exposures Exposures, both occupational and social, may provide clues to likely pathogens. When relevant, inquiries about exposure to ill contacts, animals, insects, and water should be included in the history, along with sites of residence and travel.
Other Host Factors Age, renal and hepatic function, and comorbid conditions are all considerations in the choice of and schedule for therapy. Dose adjustments should be made accordingly. In patients with decreased or unreliable oral absorption, IV therapy may be preferred to ensure adequate blood levels of drug and delivery of the antibacterial agent to the site of infection.
DURATION OF THERAPY
Whether empirical or directed, the duration of therapy should be planned in most clinical situations. Guidelines that synthesize available literature and expert opinion provide recommendations on therapy duration that are based on infecting organism, organ system, and patient factors. For example, the American Heart Association has published guidelines endorsed by the Infectious Diseases Society of America (IDSA) on diagnosis, antibacterial therapy, and management of complications of infective endocarditis. Similar guidelines from the IDSA exist for bacterial meningitis, catheter-associated urinary tract infections, intraabdominal infections, community- and hospital-acquired pneumonia, and other infections.
FAILURE OF THERAPY
If a patient does not respond to therapy, investigations often should include the collection of additional specimens for microbiologic testing and imaging as indicated. Failure to respond can be the result of an antibacterial regimen that does not address the underlying causative organism, the development of resistance during therapy, or the existence of a focus of infection at a site poorly penetrated by systemic therapy. Some infections may also require surgical interventions for cure (e.g., large abscesses, myonecrosis). Fever due to allergic drug reactions can sometimes complicate assessment of the patient’s response to antibacterial treatment.
EXPERT GUIDANCE
Selected websites with the most up-to-date information and guidance for the clinician include the following:
• Johns Hopkins ABX Guide (www.hopkins-abxguide.org)
• IDSA Practice Guidelines (www.idsociety.org/IDSA_Practice_Guidelines/)
• Center for Disease Dynamics, Economics and Policy Resistance Map (www.cddep.org/map)
• CDC Antibiotic/Antimicrobial Resistance (www.cdc.gov/drugresistance/)
CLINICAL USE OF ANTIBACTERIAL AGENTS
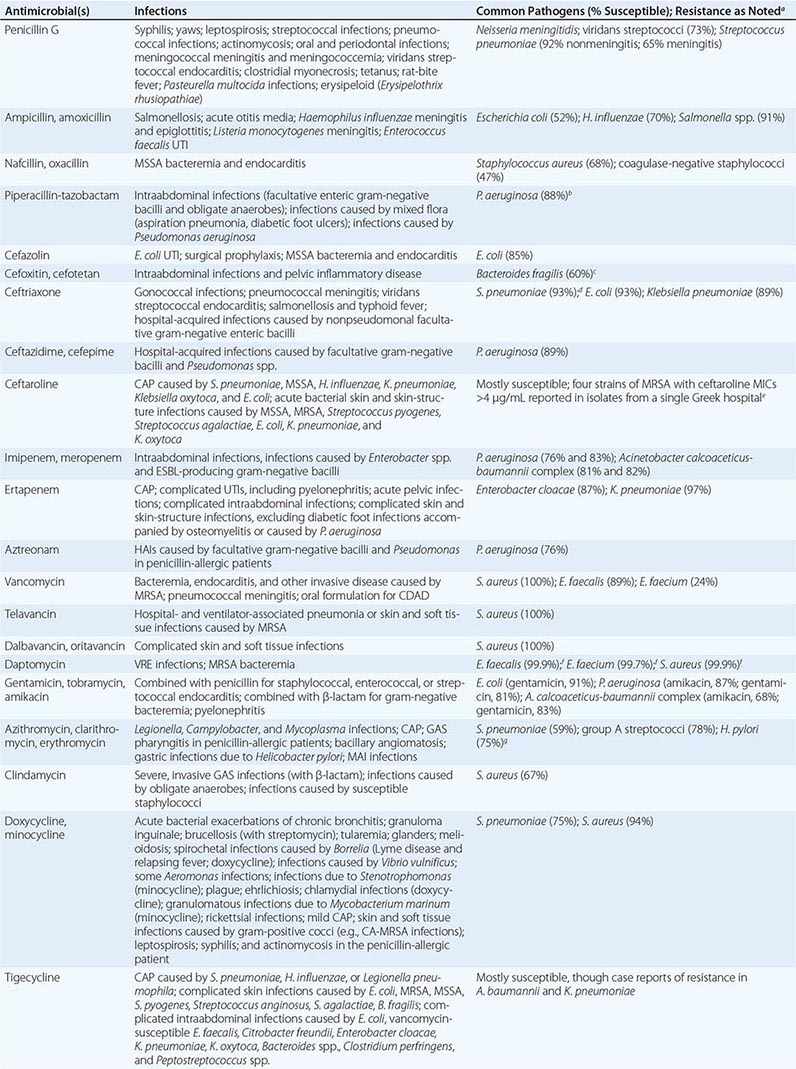
The clinical application of antibacterial therapy is guided by the spectrum of the agent and the suspected or known target pathogen. Infections for which specific antibacterial agents are among the drugs of choice are listed, along with associated pathogens and susceptibility data, in Table 170-5. Resistance rates of specific organisms are dynamic and should be taken into account in the approach to antibacterial therapy. While national resistance rates can serve as a reference, the most useful reference for the clinician is the most recent local laboratory antibiogram, which provides details on local resistance patterns, often on an annual or semiannual basis.
DRUG INDICATIONS FOR SPECIFIC INFECTIONS, ASSOCIATED PATHOGENS, AND SAMPLE SUSCEPTIBILITY RATES |
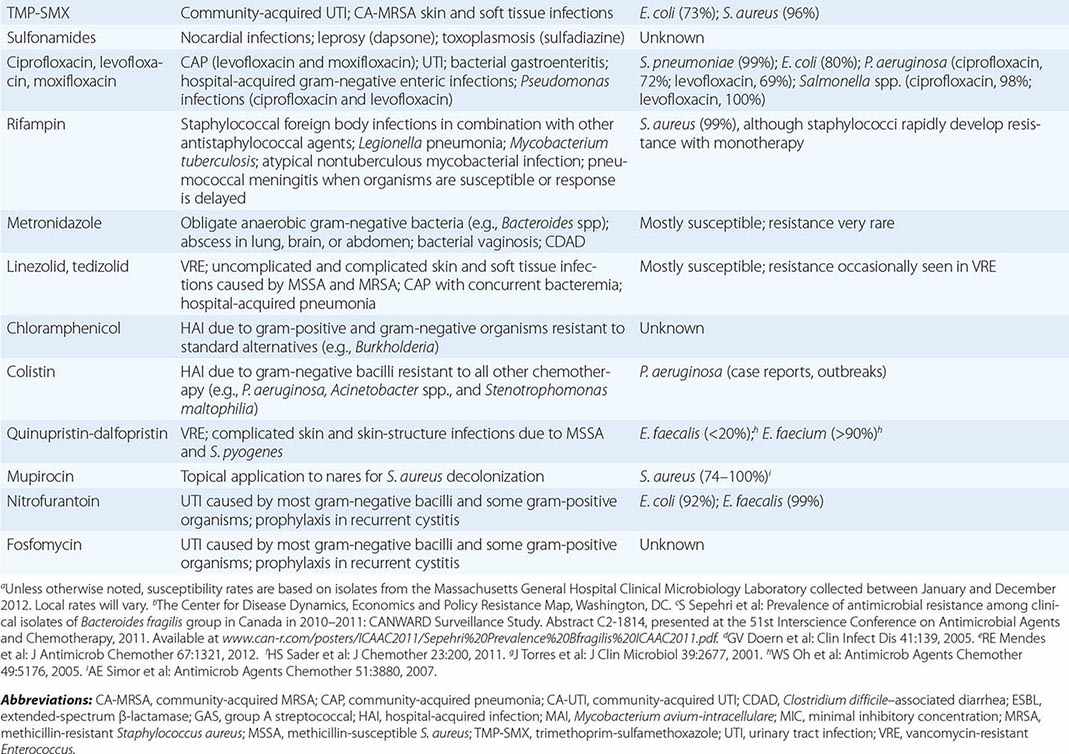
β-LACTAMS
The β-lactam class of antibiotics consists of penicillins, cephalosporins, carbapenems, and monobactams. The term β-lactam reflects the drugs’ four-membered lactam ring, which is their core structure. The differing side chains among the agents of this family determine the spectrum of activity. All β-lactams exert a bactericidal effect by inhibiting bacterial cell-wall synthesis. The β-lactams are classified as time-dependent killing agents; therefore, their clinical efficacy is best correlated with the proportion of the dosing interval during which the drug levels remain above the MIC for the pathogenic organism.
Penicillins and β-Lactamase Inhibitors Penicillin, the first β-lactam, was discovered in 1928 by Alexander Fleming. Natural penicillins, such as penicillin G, are active against non-β-lactamase-producing gram-positive and gram-negative bacteria, anaerobes, and some gram-negative cocci. Penicillin G is used for penicillin-susceptible streptococcal infections, pneumococcal and meningococcal meningitis, enterococcal endocarditis, and syphilis. The antistaphyloccocal penicillins, which have potent activity against methicillin-susceptible S. aureus (MSSA), include nafcillin, oxacillin, dicloxacillin, and flucloxacillin. Aminopenicillins, such as ampicillin and amoxicillin, provide added coverage beyond penicillin against gram-negative cocci, such as Haemophilus influenzae, and some Enterobacteriaceae, including E. coli, Proteus mirabilis, Salmonella, and Shigella. The aminopenicillins are hydrolyzed by many common β-lactamases. These drugs are commonly used for otitis media, respiratory tract infections, intraabdominal infections, endocarditis, meningitis, and urinary tract infections. The antipseudomonal penicillins include ticarcillin and piperacillin. These penicillin groups generally offer adequate anaerobic coverage; the exceptions are Bacteroides species (such as Bacteroides fragilis), which produce β-lactamases and are generally resistant. The rising prevalence of β-lactamase-producing bacteria has led to the increased use of β-lactam/β-lactamase inhibitor combinations, such as ampicillin-sulbactam, amoxicillin-clavulanate, ticarcillin-clavulanate, and piperacillin-tazobactam. The β-lactamase inhibitors themselves do not have antibacterial activity (with the exception of sulbactam, which has activity against Acinetobacter baumannii) but typically inhibit the S. aureus class A β-lactamase, β-lactamases of H. influenzae and Bacteroides species, and a number of plasmid-encoded β-lactamases. These combination agents are typically used when broader-spectrum coverage is needed—e.g., in pneumonia and intraabdominal infections. Piperacillin-tazobactam is a useful agent for broad coverage in febrile neutropenic patients. The combination agents, however, are not effective against organisms that produce AmpC β-lactamases or carbapenemases.
Cephalosporins The cephalosporin drug class encompasses five generations determined by spectrum of antibacterial activity. The first generation (cefazolin, cefadroxil, cephalexin) largely has activity against gram-positive bacteria, with some additional activity against E. coli, P. mirabilis, and K. pneumoniae. First-generation cephalosporins are commonly used for infections caused by MSSA and streptococci (e.g., skin and soft tissue infections). Cefazolin is a popular choice for surgical prophylaxis against skin organisms. The second generation (cefamandole, cefuroxime, cefaclor, cefprozil, cefuroxime axetil, cefoxitin, cefotetan) has additional activity against H. influenzae and Moraxella catarrhalis. Cefoxitin and cefotetan have potent activity against anaerobes as well. Second-generation cephalosporins are used to treat community-acquired pneumonia because of their activity against S. pneumoniae, H. influenzae, and M. catarrhalis. They are also used for other mild or moderate infections, such as acute otitis media and sinusitis. The third-generation cephalosporins are characterized by greater potency against gram-negative bacilli and reduced potency against gram-positive cocci. These cephalosporins, which include cefoperazone, cefotaxime, ceftazidime, ceftriaxone, cefdinir, cefixime, and cefpodoxime, are used for infections caused by Enterobacteriaceae, although resistance is an increasing concern. It is noteworthy that ceftazidime is the only third-generation cephalosporin with activity against P. aeruginosa but lacks activity against gram-positive bacteria. This drug is frequently used for pulmonary infections in cystic fibrosis and febrile neutropenia. Ceftriaxone penetrates the CSF and can be used to treat meningitis caused by H. influenzae, N. meningitidis, and susceptible strains of S. pneumoniae. It is also used for the treatment of later-stage Lyme disease. The fourth generation includes cefepime and cefpirome, broad-coverage agents that provide potent activity against both gram-negative bacilli, including P. aeruginosa, and gram-positive cocci. The fourth generation has clinical applications similar to those of the third generation and can be used in bacteremia, pneumonia, skin and soft tissue infections, and urinary tract infections caused by susceptible bacteria. Cefepime is also commonly used in febrile neutropenia. Ceftaroline, a fifth-generation cephalosporin, differs from the other cephalosporins in its added activity against MRSA, which is resistant to all other β-lactams. Ceftaroline’s gram-negative activity is similar to that of the third-generation cephalosporins but does not include P. aeruginosa. Ceftaroline is efficacious in community-acquired pneumonia and skin infections, but few data are available on its use for more serious infections, such as bacteremia.
Carbapenems With a few exceptions for cefepime, all penicillins and cephalosporins are ineffective in the presence of ESBLs. Carbapenems, including doripenem, imipenem, meropenem, and ertapenem, offer the most reliable coverage for strains containing ESBLs. All carbapenems have broad activity against gram-positive cocci, gram-negative bacilli, and anaerobes. None is active against MRSA, but all are active against MSSA, Streptococcus species, and Enterobacteriaceae. Ertapenem is the only carbapenem that has poor activity against P. aeruginosa and Acinetobacter. Imipenem is active against penicillin-susceptible Enterococcus faecalis but not Enterococcus faecium. Carbapenems are not active against Enterobacteriaceae containing carbapenemases. Stenotrophomonas maltophilia and some Bacillus species are intrinsically resistant to carbapenems because of a zinc-dependent carbapenemase.
Monobactams Aztreonam is the sole monobactam. Its activity is limited to gram-negative bacteria and includes P. aeruginosa and most other Enterobacteriaceae. This drug is inactivated by ESBLs and carbapenemases. The principal use for aztreonam is as an alternative to penicillins, cephalosporins, or carbapenems in patients with serious β-lactam allergy. Aztreonam is structurally related to ceftazidime and should be used cautiously in individuals with a serious ceftazidime allergy. It is commonly used in febrile neutropenia and intraabdominal infections. Aztreonam does not penetrate the CSF and should not be used for treatment of meningitis.
Adverse Reactions to β-Lactam Drugs Agents within the β-lactam class are known for several adverse effects. Gastrointestinal side effects, mainly diarrhea, are common, but hypersensitivity reactions constitute the most common adverse effect of β-lactams. The reactions’ severity can range from rash to anaphylaxis, but the rate of true anaphylactic reactions is only 0.05%. An individual with an accelerated IgE-mediated reaction to one β-lactam agent may still receive another agent within the class, but caution should be taken to choose a β-lactam that has a dissimilar side chain and a low level of cross-reactivity. For example, the second-, third-, and fourth-generation cephalosporins and the carbapenems display very low cross-reactivity in patients with penicillin allergy. Aztreonam is the only β-lactam that has no cross-reactivity with the penicillin group. In cases of severe allergy, desensitization (a graded challenge) to the indicated β-lactam, with close monitoring, may be warranted if other antibacterial options are not suitable.
β-Lactams can rarely cause serum sickness, Stevens-Johnson syndrome, nephropathy, hematologic reactions, and neurotoxicity. Neutropenia appears to be related to high doses or prolonged use. Neutropenia and interstitial nephritis caused by β-lactams generally resolve upon discontinuation of the agent. Imipenem and cefepime are associated with an increased risk of seizure, but this risk is likely a class effect and related to high doses or doses that are not adjusted in renal impairment.
GLYCOPEPTIDES
The glycopeptide antibiotics include vancomycin and telavancin. Vancomycin has activity against staphylococci (including MRSA and coagulase-negative staphylococci), streptococci (including S. pneumoniae), and enterococci. It is not active against gram-negative organisms. Vancomycin also displays activity against Bacillus species, Corynebacterium jeikeium, Listeria monocytogenes, and gram-positive anaerobes such as Peptostreptococcus, Actinomyces, Clostridium, and Propionibacterium species. Vancomycin has several important clinical uses. It is used for serious infections caused by MRSA, including health care–associated pneumonia, bacteremia, osteomyelitis, and endocarditis. It is also commonly used for skin and soft tissue infections. Oral vancomycin is not absorbed systemically and is reserved for the treatment of Clostridium difficile infection. Vancomycin is also an alternative for the treatment of infections caused by MSSA in patients who cannot tolerate β-lactams. Resistance to vancomycin is a rising concern. Strains of vancomycin-intermediate S. aureus (VISA) and vancomycin-resistant enterococci (VRE) are not uncommon. Vancomycin appears to be a concentration-dependent killer, with AUC/MIC ratio being the best predictor of efficacy (Fig. 170-2). Guidelines recommend targeting a vancomycin trough level of 15–20 μg/mL in MRSA infections in order to maintain an AUC/MIC ratio >400. When using vancomycin, clinicians should monitor for nephrotoxicity. The risk increases when trough levels are >20 μg/mL. Concomitant therapy with other nephrotoxic agents, such as aminoglycosides, also increases the risk of nephrotoxicity. Ototoxicity was reported with early formulations of vancomycin but is currently uncommon because purer formulations are available. Both of these adverse effects are reversible upon discontinuation of vancomycin. Clinicians should be aware of the “red man syndrome,” a common reaction that presents as a rapid onset of erythematous rash or pruritus on the head, face, neck, and upper trunk. This reaction is caused by histamine release from basophils and mast cells and can be treated with diphenhydramine and slowing of the vancomycin infusion.
Telavancin, dalbavancin, and oritavancin are structurally similar to vancomycin and are referred to as lipoglycopeptides. They have antibacterial activity against S. aureus (including MRSA and some strains of VISA and vancomycin-resistant S. aureus [VRSA]), streptococci, and enterococci. They also have good activity against anaerobic gram-positive organisms except for Lactobacillus and some Clostridium species. The clinical efficacy of telavancin has been demonstrated in both skin and soft tissue infections and nosocomial pneumonia, and the efficacy of dalbavancin and oritivancin has been shown in skin and soft tissue infections. The vancomycin resistance phenotype may reduce the potency of all three lipoglycopeptides, but the rate of resistance to these drugs among S. aureus and enterococci has been low. Adverse effects of telavancin include insomnia, a metallic taste, nephrotoxicity, and gastrointestinal side effects. Clinicians should be aware of the potential for electrocardiographic QTc prolongation that can increase the risk of cardiac arrhythmias when telavancin is used concomitantly with other QTc-prolonging agents. Telavancin may interfere with certain coagulation tests (e.g., causing false elevations in prothrombin time). Dalbavancin and oritavancin have safety profiles similar to that of vancomycin.
LIPOPEPTIDES
Daptomycin is a lipopeptide antibiotic with activity against a broad range of gram-positive organisms. This drug is active against staphylococci (including MRSA and coagulase-negative staphylococci), streptococci, and enterococci. Daptomycin remains active against enterococci that are resistant to vancomycin. In addition, it exhibits activity against Bacillus, Corynebacterium, Peptostreptococcus, and Clostridium species. Daptomycin’s pharmacodynamic parameter for efficacy is concentration-dependent killing. Resistance to daptomycin is rare, but MICs may be higher for VISA strains. Daptomycin is efficacious in skin and soft tissue infections, bacteremia, endocarditis, and osteomyelitis. It is an important alternative for MRSA and other gram-positive infections when bactericidal therapy is needed and vancomycin cannot be used. Daptomycin is generally well tolerated, and its main toxicity consists of elevation of creatinine phosphokinase (CPK) levels and myopathy. CPK should be monitored during daptomycin treatment, and the drug should be discontinued if muscular toxicities occur. There have also been case reports of reversible eosinophilic pneumonia associated with daptomycin use.
AMINOGLYCOSIDES
The aminoglycosides are a class of antibacterial agents with concentration-dependent activity against most gram-negative organisms. The most commonly used aminoglycosides are gentamicin, tobramycin, and amikacin, although others, such as streptomycin, kanamycin, neomycin, and paromomycin, may be used in special circumstances. Aminoglycosides have a significant dose-dependent postantibiotic effect, meaning that they have an antibacterial effect even after serum drug levels are undetectable. The postantibiotic effect and concentration-dependent killing form the rationale behind extended-interval aminoglycoside dosing, in which a larger dose is given once daily rather than smaller doses multiple times daily. Aminoglycosides are active against gram-negative bacilli, such as Enterobacteriaceae, P. aeruginosa, and Acinetobacter. They also enhance the activity of cell wall–active agents such as β-lactams or vancomycin in some gram-positive bacteria, including staphylococci and enterococci. This combination therapy is termed synergistic because the effect of both agents provides a killing effect greater than would be predicted from the effects of either agent alone. Amikacin and streptomycin have activity against Mycobacterium tuberculosis, and amikacin has activity against Mycobacterium avium-intracellulare. The aminoglycosides do not have activity against anaerobes, S. maltophilia, or Burkholderia cepacia. Aminoglycosides are used in clinical practice in a variety of infections caused by gram-negative organisms, including bacteremia and urinary tract infections. They are frequently used alone or in combination for the treatment of P. aeruginosa infection. When used in combination with a cell wall–active agent, gentamicin and streptomycin are also important for the treatment of gram-positive bacterial endocarditis. All aminoglycosides can cause nephrotoxicity and ototoxicity. The risk of nephrotoxicity is related to the dose and duration of therapy as well as the concomitant use of other nephrotoxic agents. Nephrotoxicity is usually reversible, but ototoxicity can be irreversible.
MACROLIDES AND KETOLIDES
The macrolides (azithromycin, clarithromycin, erythromycin) and ketolides (telithromycin) are classes of antibiotics that inhibit protein synthesis. Compared with erythromycin (the older antibiotic), azithromycin and clarithromycin have better oral absorption and tolerability. Azithromycin, clarithromycin, and telithromycin all have broader spectra of activity than erythromycin, which is less frequently used. These agents are commonly used in the treatment of upper and lower respiratory tract infections caused by S. pneumoniae, H. influenzae, M. catarrhalis, and atypical organisms (e.g., Chlamydia pneumoniae, Legionella pneumophila, and Mycoplasma pneumoniae); group A streptococcal pharyngitis in penicillin-allergic patients; and nontuberculous mycobacterial infections (e.g., caused by M. marinum and M. chelonae) as well as in the prophylaxis and treatment of M. avium-intracellulare infection in patients with HIV/AIDS and in combination therapy for H. pylori infection and bartonellosis. Enterobacteriaceae, Pseudomonas species, and Acinetobacter species are intrinsically resistant to macrolides as a result of decreased membrane permeability, although azithromycin is active against gram-negative diarrheal pathogens. The major adverse effects of this drug class include nausea, vomiting, diarrhea and abdominal pain, prolongation of QTc interval, exacerbation of myasthenia gravis, and tinnitus. Azithromycin specifically has been associated with an increased risk of death, especially among patients with underlying heart disease, because of the risk of QTc interval prolongation and torsades de pointes. Erythromycin, clarithromycin, and telithromycin inhibit the CYP3A4 hepatic drug-metabolizing enzyme and can result in increased levels of coadministered drugs, including benzodiazepines, statins, warfarin, cyclosporine, and tacrolimus. Azithromycin does not inhibit CYP3A4 and lacks these drug–drug interactions.
CLINDAMYCIN
Clindamycin is a lincosamide antibiotic and is bacteriostatic against some organisms and bactericidal against others. It is used most often to treat bacterial infections caused by anaerobes (e.g., B. fragilis, Clostridium perfringens, Fusobacterium species, Prevotella melaninogenicus, and Peptostreptococcus species) and susceptible staphylococci and streptococci. Clindamycin is used for treatment of dental infections, anaerobic lung abscess, and skin and soft tissue infections. It is used together with bactericidal agents (penicillins or vancomycin) to inhibit new toxin synthesis in the treatment of streptococcal or staphylococcal toxic shock syndrome. Other uses include treatment of infections caused by Capnocytophaga canimorsus, a component of combination therapy for malaria and babesiosis, and therapy for toxoplasmosis. Clindamycin has excellent oral bioavailability. Adverse effects include nausea, vomiting, diarrhea, C. difficile–associated diarrhea and pseudomembranous colitis, maculopapular rash, and (rarely) Stevens-Johnson syndrome.
TETRACYCLINES AND GLYCYLCYCLINES
The tetracyclines (doxycycline, minocycline, and tetracycline) and the glycylcyclines (tigecycline) inhibit protein synthesis and are bacteriostatic. These drugs have wide clinical uses. They are used in the treatment of skin and soft tissue infections caused by gram-positive cocci (including MRSA), spirochetal infections (e.g., Lyme disease, syphilis, leptospirosis, and relapsing fever), rickettsial infections (e.g., Rocky Mountain spotted fever), atypical pneumonia, sexually transmitted infections (e.g., Chlamydia trachomatis infection, lymphogranuloma venereum, and granuloma inguinale), infections with Nocardia and Actinomyces, brucellosis, tularemia, Whipple’s disease, and malaria. Tigecycline, the only approved agent in the glycylcycline class, is a derivative of minocycline and is indicated in the treatment of infections due to MRSA, vancomycin-sensitive enterococci, many Enterobacteriaceae, and Bacteroides species. Tigecycline has no activity against P. aeruginosa. It has been used in combination with colistin for the treatment of serious infections with multidrug-resistant gram-negative organisms. A pooled analysis of 13 clinical trials found an increased risk of death and treatment failure among patients treated with tigecycline alone. Tetracyclines have reduced absorption when coadministered with calcium- and iron-containing compounds, including milk, and doses should be spaced at least 2 h apart. The major adverse reactions to both of these classes are nausea, vomiting, diarrhea, and photosensitivity. Tetracyclines have been associated with fetal bone-growth abnormalities and should be avoided during pregnancy and in the treatment of children <8 years old.
TRIMETHOPRIM-SULFAMETHOXAZOLE
Trimethoprim-sulfamethoxazole (TMP-SMX) is an antibiotic whose two components both inhibit folate synthesis and produce antibacterial activity. TMP-SMX is active against gram-positive bacteria such as staphylococci and streptococci; however, its use against MRSA is usually limited to community-acquired infections, and its activity against Streptococcus pyogenes may not be reliable. TMP-SMX is also active against many gram-negative bacteria, including H. influenzae, E. coli, P. mirabilis, N. gonorrhoeae, and S. maltophilia. TMP-SMX does not have activity against anaerobes or P. aeruginosa. It has many uses because of its wide spectrum of activity and high oral bioavailability. Urinary tract infections, skin and soft tissue infections, and respiratory tract infections are among the common uses. Another important indication is for both prophylaxis and treatment of Pneumocysitis jirovecii infections in immunocompromised patients. Resistance to TMP-SMX has limited its use against many Enterobacteriaceae. Resistance rates among urinary isolates of E. coli are almost 25% in the United States. The most common adverse reactions associated with TMP-SMX are gastrointestinal effects such as nausea, vomiting, and diarrhea. In addition, rash is a common allergic reaction and may preclude the subsequent use of other sulfonamides. With prolonged use, leukopenia, thrombocytopenia, and granulocytopenia can develop. TMP-SMX can also cause nephrotoxicity, hyperkalemia, and hyponatremia, which are more common at high doses. TMP-SMX has several important interactions with other drugs (Table 170-4), including warfarin, phenytoin, and methotrexate.
FLUOROQUINOLONES
The fluoroquinolones include norfloxacin, ciprofloxacin, ofloxacin, levofloxacin, moxifloxacin, and gemifloxacin. Ciprofloxacin and levofloxacin have the broadest spectrum of activity against gram-negative bacteria, including P. aeruginosa (similar to that of third-generation cephalosporins). Because of the risk of selection of resistance during fluoroquinolone treatment of serious pseudomonal infections, these agents are usually used in combination with an antipseudomonal β-lactam. Levofloxacin, moxifloxacin, and gemifloxacin have additional gram-positive activity, including that against S. pneumoniae and some strains of MSSA, and are used for treatment of community-acquired pneumonia. Strains of MRSA are commonly resistant to all fluoroquinolones. Moxifloxacin is used as one component of second-line regimens for multidrug-resistant tuberculosis. Fluoroquinolones exhibit concentration-dependent killing, are well absorbed orally, and have elimination half-lives that usually support once- or twice-daily dosing. Oral coadministration with compounds containing high concentrations of aluminum, magnesium, or calcium can reduce fluoroquinolone absorption. Their penetration into prostate tissue supports their use for bacterial prostatitis. Fluoroquinolones are generally well tolerated but can cause CNS stimulatory effects, including seizures; glucose dysregulation; and tendinopathy associated with Achilles tendon rupture, particularly in older patients, organ transplant recipients, and patients taking glucocorticoids. Worsening of myasthenia gravis also has been associated with quinolone use. Moxifloxacin causes modest prolongation of the QTc interval and should be used with caution in patients receiving other QTc-prolonging drugs.
RIFAMYCINS
The rifamycins include rifampin, rifabutin, and rifapentine. Rifampin is the most commonly used rifamycin. For almost all therapeutic indications, it is used in combination with other agents to reduce the likelihood of selection of high-level rifampin resistance. Rifampin is used foremost in the treatment of mycobacterial infections—specifically, as a mainstay of combination therapy for M. tuberculosis infection or as a single agent in the treatment of latent M. tuberculosis infection. In addition, it is often used in the treatment of nontuberculous mycobacterial infection. Rifampin is used in combination regimens for the treatment of staphylococcal infections, particularly prosthetic valve endocarditis and bone infections with retained hardware. It is a component of combination therapy for brucellosis (with doxycycline) and leprosy (with dapsone for tuberculoid leprosy and with dapsone and clofazimine for lepromatous disease). Rifampin can be used alone for prophylaxis in close contacts of patients with H. influenzae or N. meningitidis meningitis. The drug has high oral bioavailability, which is further enhanced when it is taken on an empty stomach. Rifampin has several adverse effects, including elevated aminotransferase levels (14%), rash (1–5%), and gastrointestinal events such as nausea, vomiting, and diarrhea (1–2%). Its many clinically relevant interactions with other drugs mandate the clinician’s careful review of the patient’s medications before rifampin initiation to assess safety and the need for additional monitoring.
METRONIDAZOLE
Metronidazole is used in the treatment of anaerobic bacterial infections as well as infections caused by protozoa (e.g., amebiasis, giardiasis, trichomoniasis). It is the agent of choice as a component of combination therapy for polymicrobial abscesses in the lung, brain, or abdomen, the etiology of which often includes anaerobic bacteria, and for bacterial vaginosis, pelvic inflammatory disease, mild to moderate C. difficile–associated diarrhea, and anaerobic infections, such as those due to Bacteroides, Fusobacterium, and Prevotella species. Metronidazole is bactericidal against anaerobic bacteria and exhibits concentration-dependent killing. It has high oral bioavailability and tissue penetration, including penetration of the blood–brain barrier. The majority of Actinomyces, Propionibacterium, and Lactobacillus species are intrinsically resistant to metronidazole. The major adverse effects include nausea, diarrhea, and a metallic taste. Concomitant ingestion of alcohol may result in a disulfiram-like reaction, and patients are usually instructed to avoid alcohol during treatment. Long-term treatment carries the risk of leukopenia, neutropenia, peripheral neuropathy, and central nervous system toxicity manifesting as confusion, dysarthria, ataxia, nystagmus, and ophthalmoparesis. Through metronidazole’s effect on the CYP2C9 drug-metabolizing enzyme, its coadministration with warfarin can result in decreased metabolism and enhanced anticoagulant effects that require close monitoring. Concomitant administration of metronidazole with lithium can result in increased serum levels of lithium and associated toxicity; coadministration with phenytoin can result in phenytoin toxicity and possibly decreased levels of metronidazole.
OXAZOLIDINONES
Linezolid is a bacteriostatic agent and is indicated for serious infections due to resistant gram-positive bacteria, such as MRSA and VRE. The intrinsic resistance of gram-negative bacteria is mediated primarily by endogenous efflux pumps. Linezolid has excellent oral bioavailability. Adverse effects include myelosuppression and ocular and peripheral neuropathy with prolonged therapy. Peripheral neuropathy may be irreversible. Linezolid is a weak, reversible monoamine oxidase inhibitor, and coadministration with sympathomimetics and foods rich in tyramine should be avoided. Linezolid has been associated with serotonin syndrome when coadministered with selective serotonin-reuptake inhibitors. Tedizolid has properties similar to those of linezolid, but with lower dosing it may be less likely to cause adverse hematologic and neuropathic effects.
NITROFURANTOIN
Nitrofurantoin’s antibacterial activity results from the drug’s conversion to highly reactive intermediates that can damage DNA and other macromolecules. Nitrofurantoin is bactericidal, and its action is concentration dependent. It displays activity against a range of gram-positive bacteria, including S. aureus, Staphylococcus epidermidis, Staphylococcus saprophyticus, E. faecalis, Streptococcus agalactiae, group D streptococci, viridans streptococci, and corynebacteria, as well as gram-negative organisms, including E. coli and Enterobacter, Neisseria, Salmonella, and Shigella species. Nitrofurantoin is used primarily in the treatment of urinary tract infections and is preferred in the treatment of such infections in pregnancy. It may be used for the prevention of recurrent cystitis. Recently, there has been interest in the use of nitrofurantoin for treatment of urinary tract infections caused by ESBL-producing Enterobacteriaceae such as E. coli, although resistance has been growing in Latin America and parts of Europe. Coadministration with magnesium should be avoided because of decreased absorption, and patients should be encouraged to take the drug with food to increase its bioavailability and decrease the risk of adverse effects, which include nausea, vomiting, and diarrhea. Nitrofurantoin may also cause pulmonary fibrosis and drug-induced hepatitis. Because the risk of adverse reactions increases with age, the use of nitrofurantoin in elderly patients is not recommended. Patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency are at elevated risk for nitrofurantoin-associated hemolytic anemia.
POLYMYXINS
Colistin and polymyxin B act by disrupting cell membrane integrity and are active against the nonenteric pathogens P. aeruginosa and A. baumannii but not against Burkholderia. These drugs also exhibit activity against many Enterobacteriaceae, with the exceptions of Proteus, Providencia, and Serratia species. They lack activity against gram-positive bacteria. Polymyxins are bactericidal and are available in IV formulations. Colistimethate is converted to the active form (colistin) in plasma. Polymyxins are most often used for infections due to pathogens resistant to multiple other antibacterial agents, including urinary tract infections, hospital-acquired pneumonia, and bloodstream infections. Nebulized formulations have been used for adjunctive treatment of refractory ventilator-associated pneumonia. The most important adverse effect is dose-dependent reversible nephrotoxicity. Neurotoxicity, including paresthesias, muscle weakness, and confusion, is reversible and less common than nephrotoxicity.
QUINUPRISTIN-DALFOPRISTIN
Quinupristin-dalfopristin is a member of the streptogramin class of antibiotics and kills bacteria by inhibiting protein synthesis. The antibacterial spectrum of quinupristin-dalfopristin includes staphylococci (including MRSA), streptococci, and E. faecium (but not E. faecalis). This drug is also active against Corynebacterium species and L. monocytogenes. Quinupristin-dalfopristin is not reliably active against gram-negative organisms. It exhibits concentration-dependent killing, with an AUC/MIC ratio predicting efficacy. The clinical use of quinupristin-dalfopristin is largely for infections due to vancomycin-resistant E. faecium and other gram-positive bacterial infections. The drug has demonstrated efficacy in a variety of infections, including urinary tract infections, bone and joint infections, and bacteremia. Adverse effects associated with quinupristin-dalfopristin include infusion-related reactions, arthralgias, and myalgias. The arthralgias and myalgias may be severe enough to warrant drug discontinuation. Quinupristin-dalfopristin inhibits the CYP3A4 drug-metabolizing enzyme, with consequent drug interactions (Table 170-4).
FOSFOMYCIN
Fosfomycin is a phosphonic acid antibiotic that has greater activity in acidic environments and is excreted in its active form in the urine. Thus, its use is primarily for prophylaxis and treatment of uncomplicated cystitis. The drug is administered as a single 3-g dose that results in high urine concentrations for up to 48 h. Fosfomycin is active against S. aureus, vancomycin-susceptible and vancomycin-resistant enterococci, and a wide range of gram-negative organisms, including E. coli, Enterobacter species, S. marcescens, P. aeruginosa, and K. pneumoniae. Notably, the vast majority of ESBL-producing Enterobacteriaceae are susceptible to fosfomycin. A. baumannii and Burkholderia species are resistant. The emergence of resistance to fosfomycin has not been observed during treatment of cystitis but has been documented during treatment of respiratory tract infections and osteomyelitis. The few adverse effects that have been reported include nausea and diarrhea.
CHLORAMPHENICOL
The use of chloramphenicol is limited by its potentially serious toxicities. When other agents are contraindicated or ineffective, chloramphenicol represents an alternative treatment for infections, including meningitis caused by susceptible bacteria such as N. meningitidis, H. influenzae, and S. pneumoniae. It has also been used for the treatment of anthrax, brucellosis, Burkholderia infections, chlamydial infections, clostridial infections, erlichiosis, rickettsial infections, and typhoid fever. Adverse reactions include aplastic anemia, myelosuppression, and gray baby syndrome. Chloramphenicol inhibits the CYP2C19 and CYP3A4 drug-metabolizing enzymes and consequently increases levels of many classes of drugs.
APPROACH TO PROPHYLAXIS OF INFECTION
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


