OVERVIEW AND OBJECTIVES
Almost half of the elements listed in the periodic table have been found in the human body.1 An element is considered essential if a deficiency impairs a biochemical or functional process and replacement of the element corrects this impairment. Decreased intake, impaired absorption, increased excretion, and genetic abnormalities are all examples of conditions that could result in deficiency of one or several trace elements. The World Health Organization has established the dietary requirement for nutrients as the smallest amount of the nutrient needed to maintain optimal function and health. Any element that is not considered essential is classified as nonessential. Nonessential trace elements are of medical interest primarily because many of them are toxic.
The trace and toxic elements included in this chapter all have biochemical importance, whether minor or major. The essential trace elements are often associated with an enzyme (metalloenzyme) or another protein (metalloprotein) as a cofactor. Deficiencies typically impair one or more biochemical functions, and excess concentrations are associated with at least some degree of toxicity. Although trace elements, such as iron, copper, and zinc, are found in mg/L or part per million (ppm) concentrations, ultratrace elements, such as selenium, chromium, and manganese, are found in μg/L or part per billion (ppb) concentrations.
This chapter provides an overview of the trace and toxic elements frequently encountered in the clinical laboratory. The absorption, transport, distribution, metabolism (if relevant), and elimination will be described and related to the clinical significance of disease states or toxicity. In addition, an introduction to the most common testing methodologies is provided. No reference intervals have been included in the chapter text; however, several excellent sources for toxic element thresholds and trace element reference intervals are included in the bibliography. As always, the use of published thresholds and reference intervals as more than general guidelines must be done with caution, as variations in geographical location, testing methodologies, and population differences can often compromise their validity.
INSTRUMENTATION AND METHODS
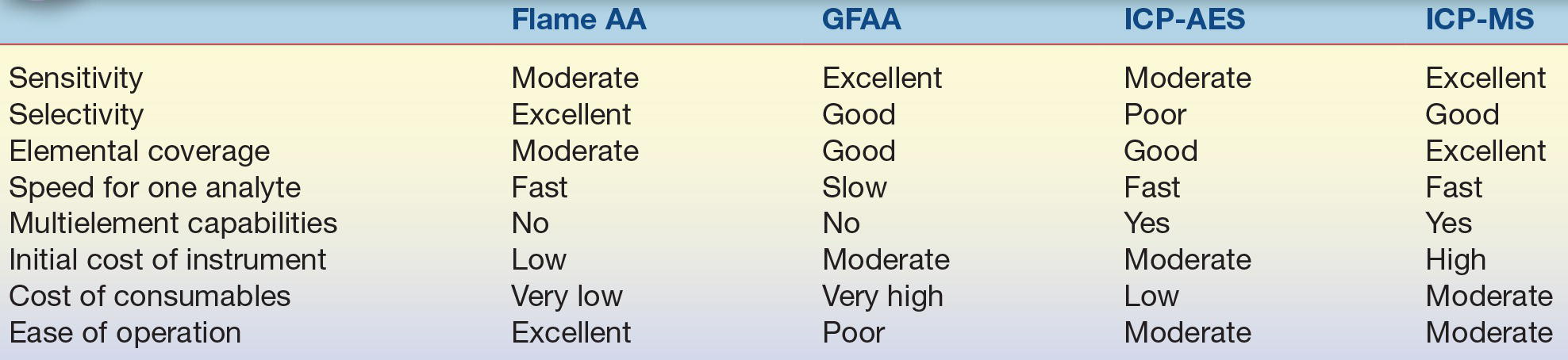
For many years, the most commonly used instrumentation for trace and toxic metal analysis has been the atomic absorption spectrometer, either with flame (FAAS) or flameless (i.e., graphite furnace, GFAAS) atomization. Atomic emission spectrometry is also useful for some elements, particularly if used in the form of inductively coupled plasma atomic emission spectroscopy (ICP-AES) for atomization and excitation. Recently, inductively coupled plasma mass spectrometry (ICP-MS) is becoming more widely used because of its sensitivity, wide range of elements covered, and relative freedom from interferences. There is no single technique that is best for all purposes. A summary of the relative advantages and disadvantages of the main techniques is given in Table 18.1.
TABLE 18.1 Relative Advantages and Disadvantages of Main Techniques for Elemental Analysis
Sample Collection and Processing
Specimens for analysis of trace elements must be collected with scrupulous attention to details such as anticoagulant, collection apparatus, and specimen type (urine, serum, plasma, or blood). Because of the low concentration in biologic specimens and the ubiquitous presence in the environment, extraordinary measures are required to prevent contamination of the specimen. This includes using special sampling and collection devices, specially cleaned glassware, and water and reagents of high purity. The selection of needles, evacuated blood collection tubes, anticoagulants and other additives, water and other reagents, pipettes, and sample cups must be carefully evaluated for use in trace and ultratrace analyses. In addition, the laboratory environment must be carefully controlled. Recommended measures include placing the trace elements laboratory in a separate room incorporating rigorous contamination control features, such as sticky mats at doors, nonshedding ceiling tiles, carefully controlled airflow to minimize particulate contamination, disposable booties worn over shoes, particle monitoring equipment, etc. Many useful measures are borrowed from those employed in semiconductor clean rooms.
Atomic Emission Spectroscopy
The simplified principle of the atomic emission spectroscopy (AES) instruments is presented in Figure 18.1.
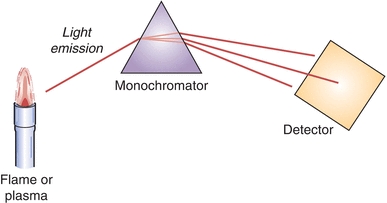
FIGURE 18.1 Simplified schematic of AES.
The three most important components of AE spectrophotometer are as follows:
1. A source, in which the sample is atomized at a sufficient temperature to produce an excited-state species. Those species will emit radiation upon relaxation back to the ground state.
2. A wavelength selecting device (monochromator), for the spectral dispersion of the radiation and separation of the analytical line from other radiation.
3. A detector permitting measurement of radiation intensity.
A liquid sample, containing element(s) of interest, is converted into an aerosol and delivered into the source, where it receives energy sufficient to emit radiation. The intensity of the emitted radiation is correlated to the concentration of an analyte and is the basis for quantitation. The most commonly used sources in AES are flame and inductively coupled plasma. Flames are capable of producing temperatures up to 3,000 K. Typical fuel gases include hydrogen and acetylene, while oxidant gases include air, oxygen, or nitrous oxide. The gases are combined in a specially designed mixing chamber. A sample is also introduced into the mixing chamber using a nebulizer that converts liquid into a fine spray. The mixing chamber and burner assembly are shown in Figure 18.2. The same assembly can be used for atomic absorption instrumentation.
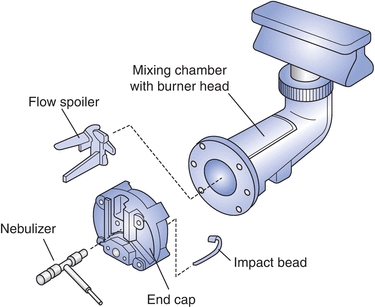
FIGURE 18.2 Mixing chamber burner for flame AA. (Courtesy of Perkin-Elmer, Waltham, MA.)
In AES, both atomic and ionic excited states can be produced (depending on the element and the source), which leads to the production of complicated emission spectra. The “emission spectrum” of an element is composed of a series of very narrow peaks (sometimes known as “lines”), with each line at a different wavelength and each line matched to a specific transition. Each element has its own characteristic emission spectrum. For example, sodium can be detected by tuning the monochromator to a wavelength of 589 nm. Ideally, each emission line of a given element would be distinct from all other emission lines of other elements. However, there are many cases where emission lines from distinct elements overlap resulting in interferences. The choice of interference-free wavelength (atomic or ionic line) may be challenging. While there are several possible wavelengths for a given element, wavelengths producing suitable analytical performance, such as limit of quantitation, freedom from interferences, robustness, etc., are selected.
The first detectors in AES used photographic film. Contemporary AES instruments feature photomultiplier tubes or array-based detector systems.
Atomic Absorption Spectroscopy
Atomic absorption spectroscopy (AAS) is an analytical procedure for the quantitative determination of elements through the absorption of optical radiation by free atoms in the gas phase. The spectra of the atoms are line spectra that are specific for the absorbing elements.
Absorption is governed by the Beer-Lambert law:
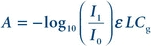 (Eq. 18-1)
(Eq. 18-1) where A is the absorbance of the sample, I0 is the incident light intensity, I1 is the transmitted light intensity, e is the molar absorptivity of the target analyte for the wavelength being used, L is the path length, and Cg is the gas-phase concentration of the target analyte. Under some simplifying assumptions, this equation takes the form:
 (Eq. 18-2)
(Eq. 18-2) where K is a constant determined by calibration, and C is the solution phase concentration of the analyte.
The simplified principle of the AAS instruments is presented in Figure 18.3.
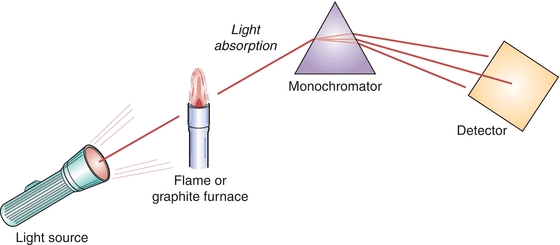
FIGURE 18.3 Simplified schematic of AAS instrumentation.
The four most important components of AA spectrophotometer are:
1. Radiation (light) source, which emits the spectrum of the analyte element
2. Atomizer, in which the atoms of the element of interest in the sample are formed
3. Monochromator, for the spectral dispersion of the radiation and separation of the analytical line from other radiation
4. Detector permitting measurement of radiation intensity
Typical radiation sources for AAS are hollow cathode lamps (HCLs) and electrodeless discharge lamps (EDLs). The HCL contains a quantity of the target element in the form of a hollow cylinder. During operation, a small quantity of the target element is vaporized, and some of the gas-phase atoms of the target element become electronically excited and emit photons with the right wavelength to be absorbed by atoms of the target element in the atomizer. While HCLs are an ideal source for determining most elements by atomic absorption, for volatile elements, the use of EDLs is recommended.
The most common sources in AAS are flame (FAAS) and graphite furnace (GFAAS, also called flameless or electrothermal AAS). The mixing chamber burner, which produces laminar flames of high optical transparency, was already described in the section on AES in this chapter. Copper, iron, and zinc are often measured by FAAS.
The graphite tubes are the most commonly used atomizers in flameless AAS. Tubes are made of high-purity polycrystalline electrographite and coated with pyrolytic graphite and can be heated to a high temperature by an electrical current. A small aliquot (usually 20 μL) of liquid sample is placed in the tube at the ambient temperature. The heating program (specifying the temperatures and times) is designed to first dry the sample, then pyrolize, vaporize, and atomize the sample, followed by a cleaning step.
Selenium, cadmium, and lead are often measured by GFAAS. GFAAS allows for measurements of both liquid and solid samples. A common problem in GFAAS is that analyte volatility depends on the molecular form of the analyte and the sample matrix. To overcome this limitation, chemical modifiers (palladium nitrate, magnesium nitrate, or a mixture of both) are frequently added to samples, calibrators, and controls.
Inductively Coupled Plasma Mass Spectrometry
Inductively coupled plasma mass spectrometry (ICP-MS) is a state-of-the-art analytical technique for elemental analysis. The term plasma in ICP refers to an ionized gas (typically argon), in which a certain proportion of electrons are free.
Like other mass spectrometers, the ICP-MS measures the mass-to-charge ratio (molecular mass divided by ionic charge [m/z]) of selected analyte ions) and includes the following components: (1) an ion source, (2) a mass analyzer, and (3) an ion detector. A simplified schematic of an ICP-MS is given in Figure 18.4.
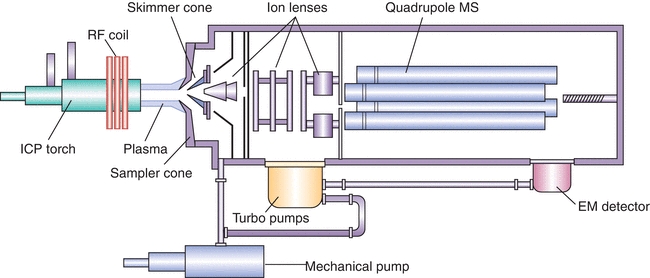
FIGURE 18.4 Simplified schematic of ICP-MS instrumentation.
The argon plasma induced by commercial ICP instruments (both ICP-AES and ICP-MS) generates temperatures ranging from 6,000 K to 10,000 K and serves several purposes. First, it dries and then vaporizes the droplets produced by the nebulizer. This step is followed by atomization of any molecular species. Finally, atoms are thermally ionized, at which point they are ready for introduction into the mass spectrometer
Nearly all ICP torches consist of three concentric quartz tubes surrounded by a coil carrying radiofrequency (RF) power. The middle tube of the torch carries the argon (Ar) that forms the plasma.
Quantitative analysis for clinical samples is best performed with the use of an internal standard. All patient samples, calibrators, and controls are diluted with an internal standard, usually a solution of an uncommon element such as yttrium. Rather than using the raw signal level of the target elements as the basis for quantitation, the signal for each of the target elements is divided by the signal of the internal standard to give signal ratios (i.e., normalized intensities).
Quadrupole Mass Spectrometers
The typical mass spectrometer used for ICP-MS is a quadrupole mass spectrometer. The analyzer consists of four parallel conducting rods arranged in a square array. Applying RF and constant (DC) voltages to the rods, the instrument can be tuned so that only ions of a specific m/z ratio can pass through the device to reach the detector. This type of instrument tends to be relatively simple to use and maintain, but the resolution (the ability to discriminate between closely spaced m/z values) is limited, being able to well resolve peaks separated by one m/z unit but not able to resolve peaks separated by a small fraction of an m/z unit.
High-Resolution Mass Spectrometers
Other ICP-MS instruments incorporate high-resolution mass spectrometers. These are usually “double focusing sector field” instruments. Such instruments separate ions of different m/z values via deflection in a magnetic field, with ions of greater m/z being deflected to a lesser degree than those of lower m/z. The magnetic field is adjusted so as to allow only ions of a selected m/z to reach the detection system at any given point in time. A second device known as an electrostatic analyzer corrects for certain nonideal effects, allowing the instrument to achieve high resolution. Commercially available high-resolution ICP-MS instruments are capable of a resolution of 10,000 (10% valley). This is enough to resolve, for example, 75As+ from 75ArCl+, both nominally 75 m/z units, but which differ by 1 × 103 units when viewed at high resolution. However, magnetic sector instruments are not able to resolve elemental isobaric interferences such as 115Sn/115In or 40Ca/40Ar, which would require resolution much higher than 10,000.
Interferences
In general, the interferences in elemental analysis are classified as spectroscopic or nonspectroscopic.
Spectroscopic
Spectral interferences generally result from a spectral overlap with the spectrum of the target analyte. For example, in AA, certain molecular species may have broad absorption spectra that may overlap the line spectra of the elements of interest, leading to false elevations of the target element concentrations. A much less common occurrence would be for the absorption spectrum of one element to overlap with that of another.
Various strategies are used to deal with spectral interferences in AAS. A continuum source background corrector may be included in the instrument design at the cost of some instrument complication. Another alternative is Zeeman background correction, which relies on shifting the atomic spectral lines by the application of a magnetic field.
In ICP-MS, spectral interferences include polyatomic species whose m/z may overlap m/z of the target analyte. For example, 56(ArO+) has the same nominal m/z as 56Fe+. The argon oxide ion, which can be a significant component of plasma generated by an ICP torch, can potentially interfere with iron analysis by ICP-MS. Another well-known polyatomic interference is argon chloride ion 75(ArCl)+ on determination of 75As+. An extensive table of polyatomic interferences in ICP-MS has been published.2
Several approaches are used to deal with polyatomic interferences in ICP-MS. One applies algebraic equations, together with relative isotopic abundance information, to mathematically correct for interferences.3 Another approach interposes a reaction cell or collision cell between the main ion lenses and the mass analyzer.4 A small amount of a gas such as helium or ammonia introduced into the cell removes interferences, either by chemical reaction or by an energy-filtering process, using the fact that polyatomic species, with their larger collisional cross sections, lose energy faster than atomic ions. Recently, a tandem mass spectrometer has been introduced to the market. Similar to conventional tandem mass spectrometers, the design includes two mass filtering quadrupoles separated by an octopole collision/reaction cell. High-resolution mass spectrometers provide a third way to remove interferences as discussed in an earlier section.
A second source of spectral interferences in ICP-MS arises from nearby elements in the periodic table. For example, tin (Sn) and cadmium (Cd) both have isotopes at 114 Da (amu), so they could potentially interfere with each other if the instrument is set to measure 114 m/z. This can usually be handled by using a different isotope for the analysis. For example, cadmium also has an isotope at 111 Da that is free from isobaric elemental interferences.
A third source of spectral interferences in ICP-MS comes from doubly charged ions. For example, 136Ba2+ appears at the same m/z as 68Zn+ (136/2 equals 68/1). These are relatively uncommon and can usually be avoided, such as by choosing a different isotope for analysis or tuning the torch to reduce multiply charged ions.
Nonspectroscopic
Matrix interferences involve the bulk physical properties of the sample to be analyzed. The aqueous samples may behave differently than organic and biological specimens, depending upon the technology used and the analyte of interest. The properties of significance are viscosity, presence of easily ionized elements, and presence of carbon. Matrix matching of the calibrators, controls, and specimens helps to overcome matrix interferences. Dilution of the specimens helps to minimize matrix effect, but it is only applicable to certain analytical techniques and to the determination of analytes with higher concentrations.
Anything that could interfere with atomization of the sample could be classified as a nonspectral interference. For example, in AA, a flame may not be hot enough for efficient atomization. Difference in sample viscosity between standards and unknown samples, resulting in differing rates of sample introduction, is another example of a nonspectral interference. In AES, anything that would prevent the efficient excitation or emission of spectral lines used for the analysis would constitute a nonspectral interference.
Elemental Speciation
The toxicity of elements may depend on their chemical forms. For example, arsenobetaine is a relatively nontoxic form of arsenic. Methylated forms of arsenic are intermediate in toxicity, and inorganic arsenic, such as As(V) and As(III), is highly toxic. In the medical evaluation of patients, it can be important to know whether an elevated arsenic level is due to relatively innocuous forms, such as arsenobetaine, perhaps from a seafood meal ingested up to 3 days before the specimen collection or by dangerous forms such as inorganic arsenic. In addition, the concentrations of methylated forms may be useful information for monitoring recovery from toxic exposure. The methodologies for elemental analysis discussed in previous sections are generally not capable of specifying the chemical form of the target elements.
So-called hyphenated techniques allow for speciation determinations. In a hyphenated analysis, the combination of two or more complementary analytical techniques is used to measure the specific form of an analyte. A classic example of this approach is liquid chromatography-ICP-MS (LC-ICP-MS). The sample is injected into a liquid chromatograph, which separates the different chemical forms of the analyte producing a characteristic retention time. Concurrently, the eluting sample is continuously analyzed by a mass spectrometer. The retention time partially identifies the analytes, and the mass spectrometer further identifies the element. In some cases, AA may substitute for MS in elemental speciation schemes. Methods for elemental speciation are becoming more common, especially in Europe. Despite clinical matrices being among the most difficult for speciation, several applications are reported; among them are the following:
Arsenic speciation in urine by LC-ICP-MS and HG-GFAAS
Copper in urine by size exclusion chromatography (SEC)-ICP-MS
Copper in red blood cells (RBCs) by SEC-ICP-MS
Lead in blood by gas chromatography (GC)-GFAAS
Selenium in serum by SEC-GFAAS
Zinc in urine by anion exchange chromatography (AEC)-ICP-MS
Alternative Analytical Techniques
Voltametric methods, such as anodic stripping voltammetry (ASV) and adsorptive stripping voltammetry (ADSV) can be used in determination of selected metals and are the basis for some point-of-care devices.5 Ion chromatography (IC) can be used for the determination of copper, iron, and zinc in blood, serum, and plasma and for the determination of zinc in urine.
Gas chromatography–mass spectrometry (GC-MS) is capable of determination of cadmium, chromium, cobalt, copper, lead, and selenium in urine, copper in serum, and lead in blood.
The methods accommodating direct analysis of solid samples, for instance, laser ablation ICP-MS (LA-ICP-MS), are gaining recognition for selected clinical applications.
ALUMINUM
Introduction
Aluminum (Al) is a crystalline silver-white ductile metal. Aluminum is the most abundant metal in the earth’s crust (~8%). It is always found combined with other elements such as oxygen, silicon, and fluorine. Aluminum as the metal is obtained from aluminum-containing minerals.
Due to its good conductivity of heat and electricity, ease of welding, tensile strength, light weight, and corrosion-resistant oxide coat, aluminum is applicable to a wide variety of industrial and household uses. Aluminum is used for beverage cans, pots and pans, airplanes, siding and roofing, and foil. Aluminum is often mixed with small amounts of other metals to form aluminum alloys, which are stronger and harder than aluminum alone.
Aluminum compounds have many different uses, for example, as alums in water treatment and alumina in abrasives and furnace linings. They are also found in consumer products such as antacids, astringents, buffered aspirin, food additives, cosmetics, and antiperspirants.
Absorption, Transport, and Excretion
The average adult in the United States ingests about 7 to 9 mg aluminum per day in food.6 The human organism can absorb aluminum and its compounds orally, by inhaling and parenterally. There is no indication of dermal absorption. Approximately 1.5% to 2% of inhaled and 0.01% to 5% of ingested aluminum are absorbed. The absorption efficiency is dependent on chemical form, particle size (inhalation), and concurrent dietary exposure to chelators such as citric acid or lactic acid.6,7
After a relatively quick uptake of aluminum into the intestinal walls, its passage into the blood is much slower. In plasma, aluminum is bound to carrier proteins such as transferrin.8 Aluminum binds to various ligands in the blood and distributes to every organ, with highest concentrations ultimately found in bone (~50% of the body burden) and lung tissues (~25% of the body burden). Aluminum levels in lungs increase with age.6
Urine accounts for 95% of aluminum excretion with 2% eliminated in the bile.
Health Effects and Toxicity
The mechanisms by which aluminum applies its toxicity are not well understood, though aluminum has been shown to interfere with a variety of enzymatic processes9 and administration of aluminum to experimental animals is known to produce encephalopathy similar to that seen in Alzheimer’s disease in man.10 Although aluminum-containing over-the-counter oral products are considered safe in healthy individuals at recommended doses, some adverse effects have been observed following long-term use in some individuals. Workers who breathe large amounts of aluminum dusts can have lung problems, such as coughing or changes that show up in chest x-rays.
Signs and symptoms of aluminum toxicity include encephalopathy (stuttering, gait disturbance, myoclonic jerks, seizures, coma, abnormal EEG), osteomalacia or aplastic bone disease (painful spontaneous fractures, hypercalcemia, and tumorous calcinosis), proximal myopathy, increased risk of infection, microcytic anemia, increased left ventricular mass, and decreased myocardial function.7
Aluminum toxicity occurs in people with renal insufficiencies who are treated by dialysis with aluminum-contaminated solutions or oral agents that contain aluminum. The clinical manifestations of aluminum toxicity include anemia, bone disease, and progressive dementia with increased concentrations of aluminum in the brain. Prolonged intravenous feeding of preterm infants with solutions containing aluminum is associated with impaired neurologic development.
Laboratory Evaluation of Aluminum Status
Aluminum is primarily measured using ICP-MS or GFAAS. Accurate measurements are often complicated by the increased risk for environmental contamination of specimens. Urine and serum levels are useful in determining toxic exposures, monitoring exposure over time, and monitoring chelation therapy.11
ARSENIC
Introduction
Arsenic (As) is a ubiquitous element displaying both metallic and nonmetallic properties. Its content in the earth’s crust is estimated at 1.5 to 2.0 mg/kg. For most people, food is the largest source of arsenic exposure (about 25 to 50 micrograms per day [μg/d]), with lower amounts coming from drinking water and air.12 Anthropogenic sources of arsenic (production of metals, burning of coil, fossil fuels, timber and its use in agriculture) release three times more of arsenic than natural sources. The main current use of arsenic is as a wood preservative. Other current and past uses of arsenic include pesticides, pigments, poison gases, ammunition manufacturing, semiconductor processing, and medicines.13
Health Effects and Toxicity
The relation of clinical signs and symptoms to arsenic exposure depends on the duration and extent of the exposure to inorganic and methylated species of arsenic, as well as the underlying clinical status of the patient. For acute arsenic exposure, the symptoms may include gastrointestinal (nausea, emesis, abdominal pain, rice water diarrhea), bone marrow (pancytopenia, anemia, basophilic stippling), cardiovascular (EKG changes), central nervous system (encephalopathy, polyneuropathy), renal (renal insufficiency, renal failure), and hepatic (hepatitis) systems. For chronic arsenic exposure, systems and symptoms may include dermatologic (Mees lines, hyperkeratosis, hyperpigmentation, alopecia), hepatic (cirrhosis, hepatomegaly), cardiovascular (hypertension, peripheral vascular disease), central nervous system (“socks and glove” neuropathy, tremor), and malignancies (squamous cell skin, hepatocellular, bladder, lung, renal). Chronic arsenic exposure has been shown to cause blackfoot disease (BFD), a severe form of peripheral vascular disease (PVD) that leads to gangrenous changes.
The white powder of arsenic trioxide is odorless, tasteless, and one of the most common poisons in human history. Doses of 0.01 to 0.05 g produce toxic symptoms. The lethal dose is reported to be between 0.12 and 0.3 g; however, recoveries from higher doses have been reported.13 Immediate treatment of expected exposure consists of lavage and use of activated charcoal to reduce arsenic absorption. The most effective antidotes for arsenic poisoning are the following chelating agents: dimercaprol (a.k.a British anti-Lewisite, BAL), penicillamine, and succimer.14
In the year 2000, the U.S. FDA approved the use of arsenic trioxide for the treatment of acute promyelocytic leukemia (APL),15 which is diagnosed in approximately 1,500 people in United States every year.
Absorption, Transport, and Excretion
The main routes of exposure are ingestion of arsenic-containing foods, water and beverages, or inhalation of contaminated air; however, arsenic toxicity is a complex topic. Organic forms of arsenic such as arsenocholine and arsenobetaine are commonly found in fish and seafood, are considered relatively nontoxic, and are cleared rapidly (1 to 2 days).16 Inorganic species of arsenic are highly toxic and occur naturally in rocks, soil, and groundwater. They are also found in many synthetic products, poisons, and industrial processes. Methylated species are intermediate in toxicity and arise primarily from metabolism of inorganic species, but small amounts may arise directly from food. Organic methylated arsenic compounds such as MMA and DMA are formed by hepatic metabolism of As(+3) and As(+5). The methylated inorganic forms are considered less toxic than As(+3) and As(+5); however, they are eliminated slowly (1 to 3 weeks). The Biological Exposure Index established by the American Conference of Governmental Industrial Hygienists (ACGIH) for the sum of inorganic and methylated metabolites of arsenic in urine is 35 μg/L. However, clinical symptoms may not be evident at 35 μg/L.14
CASE STUDY 18.1
A 27-year-old male has been seen regularly by his physician for skin lesions and increasing peripheral neuropathy over a period of several months coinciding with a home project using reclaimed wood treated with chromated copper arsenate. The initial total arsenic urine level was found to be 47 μg/L. Reflexive arsenic speciation indicated 13 μg/L As, total inorganic, 33 μg/L As, total methylated, and less than 10 μg/L of As, organic.
questions
1. Discuss the metabolism of arsenic after inorganic arsenic ingestion.
2. What is a likely cause of the elevated total arsenic result?
3. Would blood arsenic levels have helped in the diagnosis?
Laboratory Evaluation of Arsenic Status
Arsenic is primarily measured using ICP-MS, GFAAS, or HG-AAS. In most cases, arsenic is best detected by urine due to the short half-life of arsenic in blood. When arsenic speciation is desired (typically following a high total urine arsenic value reported by ICP-MS or AA), a separation method is employed either online or off-line prior to elemental analysis.
CADMIUM
Introduction
Cadmium (Cd) is a soft, bluish-white metal, which is easily cut with a knife. Principal industrial uses of cadmium include manufacture of pigments and batteries, as well as in the metal-plating and plastics industries. In the United States, the burning of fossil fuels such as coal or oil and the incineration of municipal waste materials constitute the largest sources of airborne cadmium exposure, along with zinc, lead, or copper smelters in some locations. Cadmium-containing waste products and soil contamination, primarily as a result of human activity, are becoming of concern, and the U.S. Environmental Protection Agency (EPA) has established loading rates of 20 kg/ha for high cation exchange capacity soils with a pH of 6.5 and a lower rate of 5 kg/ha for acid soils.17
Absorption, Transport, and Excretion
Based on renal function (development of proteinuria), the reference dose for cadmium in drinking water is 0.0005 mg per kg per day (mg/kg/d), and the dose for dietary exposure to cadmium is 0.001 mg/kg/d.18 Absorption of cadmium is higher in females than in males due to differences in iron stores. The absorption of inhaled cadmium in air is 10% to 50% with gastrointestinal absorption of cadmium estimated to be 5%. The absorption of cadmium in cigarette smoke is 10% to 50%, and smokers of tobacco products have about twice the cadmium abundance in their bodies as nonsmokers. For nonsmokers, the primary exposure to cadmium is through ingested food.19 Ninety percent of ingested cadmium is excreted in the feces due to the low absorbance of cadmium from the gut.20
Health Effects and Toxicity
Cadmium has no known role in normal human physiology. Toxicity is believed to be a result of protein-Cd adducts causing denaturation of the associated proteins, resulting in a loss of function.20 Ingestion of high amounts of cadmium may lead to a rapid onset with severe nausea, vomiting, and abdominal pain.20 Renal dysfunction is a common presentation for chronic cadmium exposure, often resulting in slow-onset proteinuria. Acute effects of inhalation of fumes containing cadmium include respiratory distress due to chemical pneumonitis and edema and can cause death. Breathing of cadmium vapors can also result in nasal epithelial damage and lung damage similar to emphysema.20 Cadmium exposure can affect the liver, bone, immune, blood, and nervous systems.19,21
EDTA (ethylenediaminetetraacetic acid) can be used as a chelating agent in cadmium poisoning.
Laboratory Evaluation of Cadmium Status
Cadmium is usually quantified by GFAAS and ICP-MS; ICP-AES is also used. In blood, cadmium is found mostly (70%) in the red blood cells. Cadmium in blood reflects the average uptake during the past few months and can be used for monitoring purposes but does not accurately reflect a recent exposure. Urinary excretion is about 0.001% and 0.01% of the body burden per 24 hours. At low exposure, urine cadmium reflects the total accumulation.22
CHROMIUM
Introduction
Chromium (Cr), from the Greek word chroma (“color”), makes rubies red and emeralds green. Chromium is the 21st most abundant element in the earth’s crust and is used in the manufacturing of stainless steel. Occupational exposure to chromium occurs in wood treatment, stainless steel welding, chrome plating, the leather tanning industry, and the use of lead chromate or strontium chromate paints. Chromium exists in two main valency states: trivalent and hexavalent.
Absorption, Transport, and Excretion
Cr(+6) is better absorbed and much more toxic than Cr(+3).22 Both transferrin and albumin are involved in chromium absorption and transport.23 Transferrin binds the newly absorbed chromium at site B, while albumin acts as an acceptor and transporter of chromium if the transferrin sites are saturated.24 Other plasma proteins, including β- and γ-globulins and lipoproteins, bind chromium.
Health Effects, Deficiency, and Toxicity
Cr(+3) is an essential dietary element and plays a role in maintaining normal metabolism of glucose, fat, and cholesterol. The estimated safe and adequate daily intake of chromium for adults is in the range of 50 to 200 μg/d, although data are insufficient to establish a recommended daily allowance.25
Dietary chromium deficiency is relatively uncommon, and most cases occur in persons with specific clinical situations such as total parenteral nutrition, diabetes, or malnutrition. Chromium deficiency is characterized by glucose intolerance, glycosuria, hypercholesterolemia, decreased longevity, decreased sperm counts, and impaired fertility.25
Cr(+6) compounds are powerful oxidizing agents and are more toxic systemically than Cr(+3) compounds, given similar amounts and solubilities. At physiological pH, Cr(+6) forms 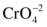 and readily passes through cell membranes due to its similarity to essential phosphate and sulfate oxyanions. Intracellularly, Cr(+6) is reduced to reactive intermediates, producing free radicals and oxidizing DNA, both potentially inducing cell death.26 Severe dermatitis and skin ulcers can result from contact with Cr(+6) salts. Up to 20% of chromium workers develop contact dermatitis. Allergic dermatitis with eczema has been reported in printers, cement workers, metal workers, painters, and leather tanners. Data suggest that a Cr(+3)–protein complex is responsible for the allergic reaction.25
and readily passes through cell membranes due to its similarity to essential phosphate and sulfate oxyanions. Intracellularly, Cr(+6) is reduced to reactive intermediates, producing free radicals and oxidizing DNA, both potentially inducing cell death.26 Severe dermatitis and skin ulcers can result from contact with Cr(+6) salts. Up to 20% of chromium workers develop contact dermatitis. Allergic dermatitis with eczema has been reported in printers, cement workers, metal workers, painters, and leather tanners. Data suggest that a Cr(+3)–protein complex is responsible for the allergic reaction.25
When inhaled, Cr(+6) is a respiratory tract irritant, resulting in airway irritation, airway obstruction, and possibly lung cancer. The target organ of inhaled chromium is the lung; the kidneys, liver, skin, and immune system may also be affected.
Low-dose, chronic chromium exposure typically results only in transient renal effects. Elevated urinary β2-microglobulin levels (an indicator of renal tubular damage) have been found in chrome platers, and higher levels have generally been observed in younger persons exposed to higher Cr(+6) concentrations.25
In May 2011, the FDA issued orders for postmarket surveillance studies to manufacturers of metal-on-metal (MoM) hip replacement systems in response to an increasing concern over failed implant devices reported in Europe.27 In the United States, no guidelines have been established for the assessment of metal ions in asymptomatic patients due to a lack of knowledge regarding the prevalence of adverse events in the US population and no clear threshold levels associated with an adverse event.
Laboratory Evaluation of Chromium Status
Chromium may be determined by GFAAS, NAA, or ICP-MS. Plasma, serum, and urine do not indicate the total body status of the individual, whereas urine levels may be useful for metabolic studies.23 In the setting of suspected failure of MoM hip implants that use a cobalt–chrome alloy femoral head, serum is the preferred specimen type for both chromium and cobalt analysis.28
COPPER
Introduction
Copper (Cu) is a relatively soft yet tough metal with excellent electrical and heat conducting properties. Copper is widely distributed in nature both in its elemental form and in compounds. Copper forms alloys with zinc (brass), tin (bronze), and nickel (cupronickel, widely used in coins). Copper is an essential trace element found in four oxidation states, Cu(0), Cu(+1), Cu(+2), and Cu(+3), with Cu(+2) the most stable of all oxidation states.29 Copper is an important cofactor for several metalloenzymes and is critical for the reduction of iron in heme synthesis.
Absorption, Transport, and Excretion
The copper content in the normal human adult is 50 to 120 mg. Copper is distributed through the body with the highest concentrations found in the liver, brain, heart, and kidneys. Hepatic copper accounts for about 10% of the total copper in the body.30 Copper is also found in the cornea, spleen, intestine, and lung. The amount of copper absorbed from the intestine is 50% to 80% of ingested copper.25 The average daily intake is approximately 10 mg or more of copper.30 The exact mechanisms by which copper is absorbed and transported by the intestine are unknown, but an active transport mechanism at low concentrations and passive diffusion at high concentrations have been proposed. Copper is transported to the liver bound to albumin, transcuprein, and low-molecular-weight components in the portal system. In the liver, copper is incorporated into ceruloplasmin for distribution throughout the body.29 Ceruloplasmin is an α2-globulin, and each 132,000-molecular-weight molecule contains six atoms of copper.30
In a normal physiological state, 98% of copper excretion is through the bile, with copper losses in the urine and sweat constituting approximately 2% of dietary intake.25 Menstrual losses of copper are minor.
Health Effects, Deficiency, and Toxicity
Copper is a component of several metalloenzymes, including ceruloplasmin, cytochrome C oxidase, superoxide dismutase, tyrosinase, metallothionein, dopamine hydroxylase, lysyl oxidase, clotting factor V, and an unknown enzyme that cross-links keratin in hair.
Copper deficiency is observed in premature infants, and copper absorption is impaired in severe diffuse diseases of small bowel, lymph sarcoma, and scleroderma.30 Copper deficiency is related to malnutrition, malabsorption, chronic diarrhea, hyperalimentation, and prolonged feeding with low-copper, total-milk diets. Signs of copper deficiency include (1) neutropenia and hypochromic anemia in the early stages, (2) osteoporosis and various bone and joint abnormalities that reflect deficient copper-dependent cross-linking of bone collagen and connective tissue, (3) decreased pigmentation of the skin and general pallor, and (4) in the later stages, possible neurologic abnormalities (hypotonia, apnea, psychomotor retardation).25
Subclinical copper depletion contributes to an increased risk of coronary heart disease. An extreme form of copper deficiency is seen in Menkes’ disease, with symptoms usually appearing at the age of 3 months and death usually occurring by the age of 5. This invariably fatal, progressive brain disease is characterized by peculiar hair, called kinky or steely, and retardation of growth. Clinical signs include progressive mental deterioration, coarse feces, disturbance of muscle tone, seizures, and episodes of severe hypothermia.
Copper toxicity has been associated with living near copper-producing facilities, using water from copper pipes or cooking with copper-lined vessels or exposure to algaecides, herbicides, pyrotechnics, ceramic glazes, electrical wiring, or welding supplies. Because of its redox potential, copper is an irritant to epithelia and mucous membranes and can cause hepatic and renal damage with hemolysis.31 Copper-induced emesis has a characteristic blue-green color.
Wilson’s disease is a genetically determined copper accumulation disease that usually presents between the ages of 6 and 40 years. Clinical findings include neurologic disorders, liver dysfunction, and Kayser-Fleischer rings (green-brown discoloration) in the cornea caused by copper deposition. Early diagnosis of Wilson’s disease is important because complications can be effectively prevented, and in some cases, the disease can be halted with use of zinc acetate or chelation therapy.25 Serum ceruloplasmin levels and the direct measurement of free copper are key diagnostic steps in the diagnosis of Wilson’s disease.32
Laboratory Evaluation of Copper Status
Copper is measured by flame AAS, ICP-MS, ICP-AES, and ASV. Serum copper and urine copper are used to monitor for nutritional adequacy and subacute management of copper toxicity. Direct measurement of free copper and ceruloplasmin in serum is used to screen for Wilson’s disease. Common trends in laboratory testing seen in various diseases states are summarized in Table 18.2.
TABLE 18.2 Interpretation of Copper Testing Results
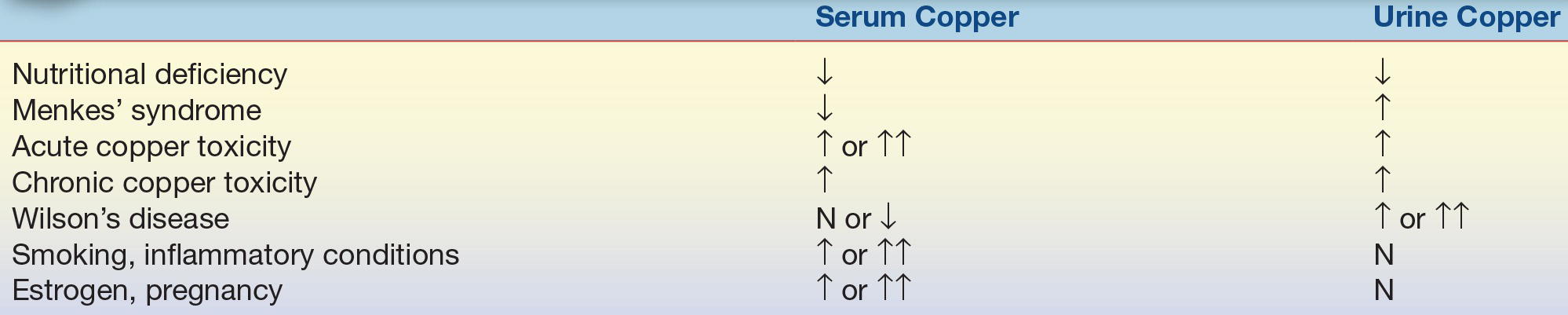
N, normal; ↓, decreased; ↑, increased; ↑↑, significantly increased. Source: Jacobs DS, DeMott WR, Oxley DK. Jacobs & Demott Laboratory Test Handbook. 5th ed. Hudson, OH: Lexi-Comp.; 2001:1031.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


