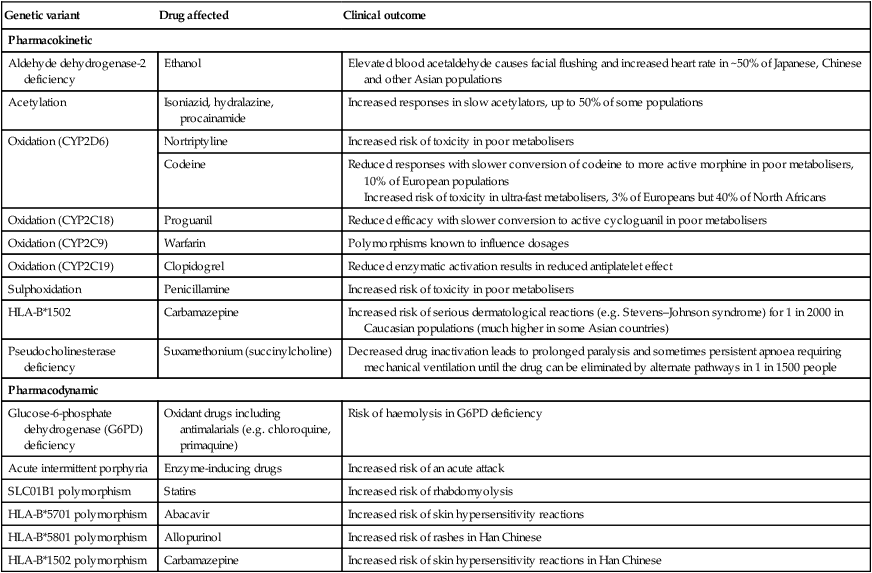
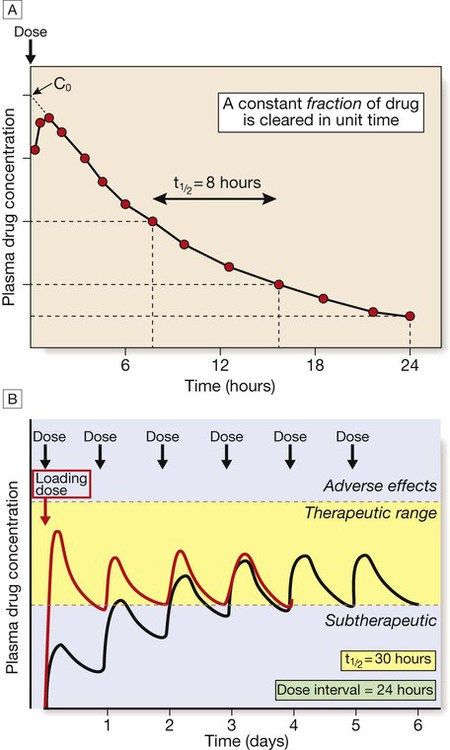
Prescribing medicines is a major tool used by most doctors to restore or preserve the health of their patients. Medicines contain drugs (the specific chemical substances with pharmacological effects), either alone or in combination, in a formulation mixed with other ingredients. The beneficial effects of medicines must be weighed against their cost and the risks of adverse drug reactions and interactions, often caused by injudicious prescribing decisions and by prescribing errors. The modern prescriber must meet the challenges posed by an increasing number of drugs and formulations available and of indications for prescribing them, and the greater complexity of treatment regimens followed by individual patients (‘polypharmacy’, a particular challenge in the ageing population). The purpose of this chapter is to elaborate on the principles and practice that underpin good prescribing (Box 2.1). • Consider factors influencing the patient’s responses to therapy (age, concomitant drug therapy, renal and liver function etc.)* • Establish the therapeutic goal* • Choose the therapeutic approach* • Choose the drug and its formulation (the ‘medicine’) • Choose the dose, route and frequency • Choose the duration of therapy • Write an unambiguous prescription (or ‘medication order’) • Inform the patient about the treatment and its likely effects • Monitor treatment effects, both beneficial and harmful *These steps in particular take the patient’s views into consideration to establish a therapeutic partnership. Prescribers need to understand what the drug does to the body (pharmacodynamics) and what the body does to the drug (pharmacokinetics) (Fig. 2.1). Although this chapter is focused on the most common drugs, which are synthetic small molecules, the same principles apply to the increasingly numerous ‘biological’ therapies (sometimes abbreviated to ‘biologics’) now in use, which include peptides, proteins, enzymes and monoclonal antibodies (p. 74). Modern drugs are usually discovered by screening compounds for activity either to stimulate or to block the function of a specific molecular target, which is predicted to have a beneficial effect in a particular disease (Box 2.2). Other drugs have useful but less selective chemical properties, such as chelators (e.g. for treatment of iron or copper overload), osmotic agents (used as diuretics in cerebral oedema) or general anaesthetics (that alter the biophysical properties of lipid membranes). The following characteristics of the interaction of drugs with receptors illustrate some of the important determinants of the effects of drugs: • Affinity describes the propensity for a drug to bind to a receptor and is related to the ‘molecular fit’ and the strength of the chemical bond. Some drug–receptor interactions are irreversible, either because the affinity is so strong or because the drug modifies the structure of its molecular target. • Selectivity describes the propensity for a drug to bind to one target rather than another. Selectivity is a relative term, not to be confused with absolute specificity. It is common for drugs targeted at a particular subtype of receptor to exhibit some effect at other subtypes. For example, β-adrenoceptors can be subtyped on the basis of their responsiveness to the endogenous agonist noradrenaline (norepinephrine): the concentration of noradrenaline required to cause bronchodilatation (via β2-adrenoceptors) is ten times higher than that required to cause tachycardia (via β1-adrenoceptors). ‘Cardioselective’ β-blockers have anti-anginal effects on the heart (β1) but may still cause bronchospasm in the lung (β2) and are contraindicated for asthmatic patients. • Agonists bind to a receptor to produce a conformational change that is coupled to a biological response. As agonist concentration increases, so does the proportion of receptors occupied, and hence the biological effect. Partial agonists activate the receptor, but cannot produce a maximal signalling effect equivalent to that of a full agonist even when all available receptors are occupied. • Antagonists bind to a receptor but do not produce the conformational change that initiates an intracellular signal. A competitive antagonist competes with endogenous ligands to occupy receptor binding sites, with the resulting antagonism depending on the relative affinities and concentrations of drug and ligand. Non-competitive antagonists inhibit the effect of an agonist by mechanisms other than direct competition for receptor binding with the agonist (e.g. by affecting post-receptor signalling). Plotting the logarithm of drug dose against drug response typically produces a sigmoidal dose–response curve (Fig. 2.2). Progressive increases in drug dose (which for most drugs is proportional to the plasma drug concentration) produce increasing response, but only within a relatively narrow range of dose; further increases in dose beyond this range produce little extra effect. The following characteristics of the drug response are useful in comparing different drugs: • Efficacy describes the extent to which a drug can produce a target-specific response when all available receptors or binding sites are occupied (i.e. Emax on the dose–response curve). A full agonist can produce the maximum response of which the receptor is capable, while a partial agonist at the same receptor will have lower efficacy. Therapeutic efficacy describes the effect of the drug on a desired biological endpoint, and can be used to compare drugs that act via different pharmacological mechanisms (e.g. loop diuretics induce a greater diuresis than thiazide diuretics and therefore have greater therapeutic efficacy). • Potency describes the amount of drug required for a given response. More potent drugs produce biological effects at lower doses, so they have a lower ED50. A less potent drug can still have an equivalent efficacy if it is given in higher doses. The dose–response relationship varies between patients because of variations in the many determinants of pharmacokinetics and pharmacodynamics. In clinical practice, the prescriber is unable to construct a dose–response curve for each individual patient. Therefore, most drugs are licensed for use within a recommended range of doses that is expected to reach close to the top of the dose–response curve for most patients. However, it is sometimes possible to achieve the desired therapeutic efficacy at doses towards the lower end of, or even below, the recommended range. The adverse effects of drugs are often dose-related in a similar way to the beneficial effects, although the dose–response curve for these adverse effects is normally shifted to the right (see Fig. 2.2). The ratio of the ED50 for therapeutic efficacy and for a major adverse effect is known as the ‘therapeutic index’. In reality, drugs have multiple potential adverse effects but the concept of therapeutic index is usually reserved for those requiring dose reduction or discontinuation. For most drugs, the therapeutic index is greater than 100 but there are some notable exceptions with therapeutic indices less than 10 (e.g. digoxin, warfarin, insulin, phenytoin, opioids). The doses of such drugs have to be titrated carefully for individual patients to maximise benefits but avoid adverse effects. • Tachyphylaxis describes desensitisation that occurs very rapidly, sometimes with the initial dose. This rapid loss of response implies depletion of chemicals that may be necessary for the pharmacological actions of the drug (e.g. a stored neurotransmitter released from a nerve terminal) or receptor phosphorylation. • Tolerance describes a more gradual loss of response to a drug that occurs over days or weeks. This slower change implies changes in receptor numbers or the development of counter-regulatory physiological changes that offset the actions of the drug (e.g. accumulation of salt and water in response to vasodilator therapy). • Drug resistance is a term normally reserved for describing the loss of effectiveness of an antimicrobial (p. 151) or cancer chemotherapy drug. • In addition to these pharmacodynamic causes of desensitisation, reduced response may be the consequence of lower plasma and tissue drug concentrations as a result of altered pharmacokinetics (see below). When drugs induce chemical, hormonal and physiological changes that offset their actions, discontinuation may allow these changes to cause ‘rebound’ withdrawal effects (Box 2.3). Understanding ‘what the body does to the drug’ (Fig. 2.3) is extremely important for prescribers because this forms the basis on which the optimal route of administration and dose regimen are chosen and explains the majority of inter-individual variation in the response to drug therapy. Absorption is the process by which drug molecules gain access to the blood stream. The rate and extent of drug absorption depend on the route of administration (see Fig. 2.3). These routes involve administration via the gastrointestinal tract: • Oral. This is the commonest route of administration because it is simple, convenient and readily used by patients to self-administer their medicines. Absorption after an oral dose is a complex process that depends on the drug being swallowed, surviving exposure to gastric acid, avoiding unacceptable food binding, being absorbed across the small bowel mucosa into the portal venous system, and surviving metabolism by gut wall or liver enzymes (‘first-pass metabolism’). As a consequence, absorption is frequently incomplete following oral administration. The term ‘bioavailability’ describes the proportion of the dose that reaches the systemic circulation intact. • Buccal, intranasal and sublingual (SL). These routes have the advantage of enabling rapid absorption into the systemic circulation without the uncertainties associated with oral administration (e.g. organic nitrates for angina pectoris, triptans for migraine, opioid analgesics). • Rectal (PR). The rectal mucosa is occasionally used as a site of drug administration when the oral route is compromised because of nausea and vomiting or unconsciousness (e.g. diazepam in status epilepticus). These routes avoid absorption via the gastrointestinal tract and first-pass metabolism in the liver: • Intravenous (IV). The IV route enables all of a dose to enter the systemic circulation reliably, without any concerns about absorption or first-pass metabolism (i.e. the dose is 100% bioavailable), and rapidly achieve a high plasma concentration. It is ideal for very ill patients when a rapid, certain effect is critical to outcome (e.g. benzylpenicillin for meningococcal meningitis). • Intramuscular (IM). IM administration is easier to achieve than the IV route (e.g. adrenaline (epinephrine) for acute anaphylaxis) but absorption is less predictable and depends on muscle blood flow. • Subcutaneous (SC). The SC route is ideal for drugs that have to be administered parenterally because of low oral bioavailability, are absorbed well from subcutaneous fat, and might ideally be injected by patients themselves (e.g. insulin, heparin). • Transdermal. A transdermal patch can enable a drug to be absorbed through the skin and into the circulation (e.g. oestrogens, testosterone, nicotine, nitrates). • Topical application of a drug involves direct administration to the site of action (e.g. skin, eye, ear). This has the advantage of achieving sufficient concentration at this site while minimising systemic exposure and the risk of adverse effects elsewhere. • Inhaled (INH) administration allows drugs to be delivered directly to a target in the respiratory tree, usually the small airways (e.g. salbutamol, beclometasone). However, a significant proportion of the inhaled dose may be absorbed from the lung or is swallowed and can reach the systemic circulation. The most common mode of delivery is the metered-dose inhaler but its success depends on some degree of manual dexterity and timing (see Fig. 19.23, p. 670). Patients who find these difficult may use a ‘spacer’ device to improve drug delivery. A special mode of inhaled delivery is via a nebulised solution created by using pressurised oxygen or air to break up solutions and suspensions into small aerosol droplets that can be directly inhaled from the mouthpiece of the device. where D is the amount of drug given and C0 is the initial plasma concentration (Fig. 2.4A). Drugs that are highly bound to plasma proteins may have a Vd below 10 L (e.g. warfarin, aspirin), while those that diffuse into the interstitial fluid but do not enter cells because they have low lipid solubility may have a Vd between 10 and 30 L (e.g. gentamicin, amoxicillin). It is an ‘apparent’ volume because those drugs that are lipid-soluble and highly tissue-bound may have a Vd of greater than 100 L (e.g. digoxin, amitriptyline). Drugs with a larger Vd are eliminated more slowly from the body. Phase II metabolism involves combining phase I metabolites with an endogenous substrate to form an inactive conjugate that is much more water-soluble. Reactions include glucuronidation, sulphation, acetylation or methylation, and conjugation with glutathione. This is necessary to enable renal excretion because lipid-soluble metabolites will simply diffuse back into the body after glomerular filtration (p. 430). Excretion is the process by which drugs and their metabolites are removed from the body. Faecal excretion is the predominant route of elimination for drugs with high molecular weight, including those that are excreted in the bile after conjugation with glucuronide in the liver, and any drugs that are not absorbed after enteral administration. Molecules of drug or metabolite that are excreted in the bile enter the small intestine, where they may, if they are sufficiently lipid-soluble, be reabsorbed through the gut wall and return to the liver via the portal vein (see Fig. 2.3). This recycling between the liver, bile, gut and portal vein is known as ‘enterohepatic circulation’ and can significantly prolong the residence of drugs in the body. For most drugs, elimination is a high-capacity process that does not become saturated, even at high dosage. The rate of elimination is therefore directly proportional to the drug concentration because of the ‘law of mass action’, whereby higher drug concentrations will drive faster metabolic reactions and support higher renal filtration rates. This results in ‘first-order’ kinetics, when a constant fraction of the drug remaining in the circulation is eliminated in a given time and the decline in concentration over time is exponential (see Fig. 2.4A). This elimination can be described by the drug’s half-life (t1/2), i.e. the time taken for the plasma drug concentration to halve, which remains constant throughout the period of drug elimination. The significance of this phenomenon for prescribers is that the effect of increasing doses on plasma concentration is predictable – a doubled dose leads to a doubled concentration at all time points. The goal of therapy is usually to maintain drug concentrations within the therapeutic range (see Fig. 2.2) over several days (e.g. antibiotics) or even for months or years (e.g. antihypertensives, lipid-lowering drugs, thyroid hormone replacement therapy). This goal is rarely achieved with single doses, so prescribers have to plan a regimen of repeated doses. This involves choosing the size of each individual dose and the frequency of dose administration. As illustrated in Figure 2.4B, the time taken to reach drug concentrations within the therapeutic range depends on the half-life of the drug. Typically, with doses administered regularly, it takes approximately 5 half-lives to reach a ‘steady state’ in which the rate of drug elimination is equal to the rate of drug administration. This applies when starting new drugs and when adjusting doses of current drugs. With appropriate dose selection, steady state drug concentrations will be maintained within the therapeutic range. This is important for prescribers because it means that the effects of a new prescription, or dose titration, for a drug with a long half-life (e.g. digoxin – 36 hours) may not be known for a few days. In contrast, drugs with a very short half-life (e.g. dobutamine – 2 minutes) have to be given continuously by infusion but reach a new steady state within minutes. Prescribers have numerous sources of guidance about how to use drugs appropriately (e.g. dose, route, frequency, duration) for many conditions. However, this advice is based on average dose–response data derived from observations in many individuals. When applying this information to an individual patient, prescribers must take account of inter-individual variability in response. Some of this variability is predictable and good prescribers are able to anticipate it and adjust their prescriptions accordingly to maximise the chances of benefit and minimise harm. Inter-individual variation in responses also mandates that effects of treatment should be monitored (p. 39). Some inter-individual variation in drug response is accounted for by differences in pharmacodynamics. For example, the beneficial natriuresis produced by the loop diuretic furosemide is often significantly reduced at a given dose in patients with renal impairment, while confusion caused by opioid analgesics is more likely in the elderly. However, differences in pharmacokinetics more commonly account for different drug responses. Examples of factors influencing the absorption, metabolism and excretion of drugs are shown in Box 2.4. It is hoped that a significant proportion of the inter-individual variation in drug responses can be explained by studying genetic differences in single genes (‘pharmacogenetics’) (Box 2.5) or the effects of multiple gene variants (‘pharmacogenomics’). The aim is to identify those patients most likely to benefit from specific treatments and those most susceptible to adverse effects. In this way, it may be possible to select drugs and dose regimens for individual patients to maximise the benefit:hazard ratio (‘personalised medicine’). Some important definitions for the adverse effects of drugs are: • Adverse event. A harmful event that occurs while a patient is taking a drug, irrespective of whether the drug is suspected of being the cause. • Adverse drug reaction (ADR). An unwanted or harmful reaction that is experienced following the administration of a drug or combination of drugs under normal conditions of use and is suspected to be related to the drug. An ADR will usually require the drug to be discontinued or the dose reduced. • Side-effect. Any effect caused by a drug other than the intended therapeutic effect, whether beneficial, neutral or harmful. The term ‘side-effect’ is often used interchangeably with ‘ADR’, although the former usually implies an effect that is less harmful, is predictable and may not require discontinuation of therapy (e.g. ankle oedema with vasodilators). • Drug toxicity. Adverse effects of a drug that occur because the dose or plasma concentration has risen above the therapeutic range, either unintentionally or intentionally (drug overdose, see Fig. 2.2, p. 19). • Drug abuse. The misuse of recreational or therapeutic drugs that may lead to addiction or dependence, serious physiological injury (such as liver damage), psychological harm (abnormal behaviour patterns, hallucinations, memory loss) or death (p. 240). ADRs are a common cause of illness, accounting in the United Kingdom (UK) for approximately 3% of consultations in primary care and 7% of emergency admissions to hospital, and affecting around 15% of hospital inpatients. Many ‘disease’ presentations are eventually attributed to ADRs, emphasising the importance of always taking a careful drug history (Box 2.6). Factors accounting for the rising prevalence of ADRs are the increasing age of patients, polypharmacy (higher risk of drug interactions), increasing availability of over-the-counter medicines, increase in use of herbal or traditional medicines, and increase in medicines available via the Internet. Risk factors for ADRs are shown in Box 2.7. Information from the patient (or carer) • Current prescribed drugs, including formulations (e.g. modified-release tablets), doses, routes of administration, frequency and timing, duration of treatment • Other medications that are often forgotten (e.g. over-the-counter drugs, herbal remedies, vitamins) • Drugs that have been taken in the recent past and reasons for stopping them • Previous drug hypersensitivity reactions, their nature and time course (e.g. rash, anaphylaxis) • Previous ADRs, their nature and time course (e.g. muscle aches with simvastatin) • Adherence to therapy (e.g. ‘are you taking your medication regularly?’) • Elderly age (e.g. low physiological reserve) • Gender (e.g. ACE inhibitor-induced cough in women) • Polypharmacy (e.g. drug interactions) • Genetic predisposition (see Box 2.5) • Hypersensitivity/allergy (e.g. β-lactam antibiotics) • Diseases altering pharmacokinetics (e.g. hepatic or renal impairment) or pharmacodynamic responses (e.g. bladder instability)
Therapeutics and good prescribing
 2.1 Steps in good prescribing
2.1 Steps in good prescribing
Principles of clinical pharmacology
Pharmacodynamics
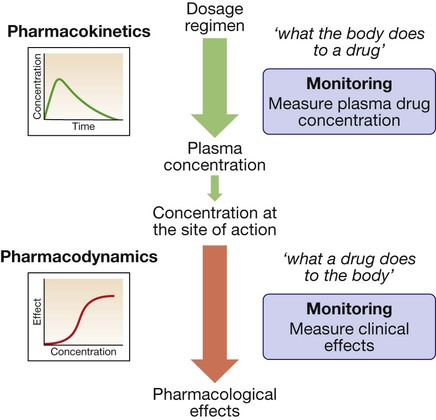
Drug targets and mechanisms of action
Dose–response relationships
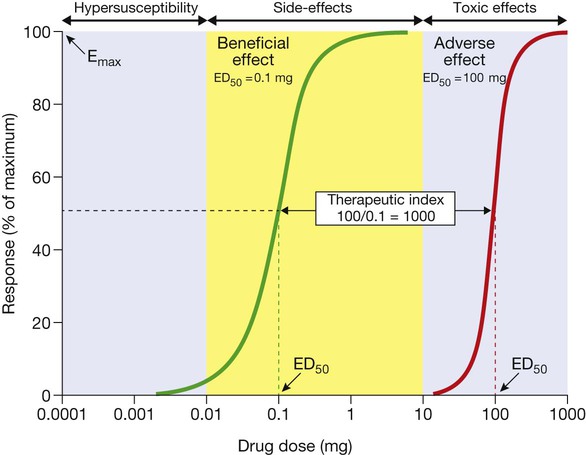
The green curve represents the beneficial effect of the drug. The maximum response on the curve is the Emax and the dose (or concentration) producing half this value (Emax/2) is the ED50 (or EC50). The red curve illustrates the dose–response relationship for the most important adverse effect of this drug. This occurs at much higher doses; the ratio between the ED50 for the adverse effect and that for the beneficial effect is the ‘therapeutic index’, which indicates how much margin there is for prescribers when choosing a dose that will provide beneficial effects without also causing this adverse effect. Adverse effects that occur at doses above the therapeutic range (yellow area) are normally called ‘toxic effects’, while those occurring within the therapeutic range are ‘side-effects’ and those below it are ‘hyper-susceptibility effects’.
Therapeutic index
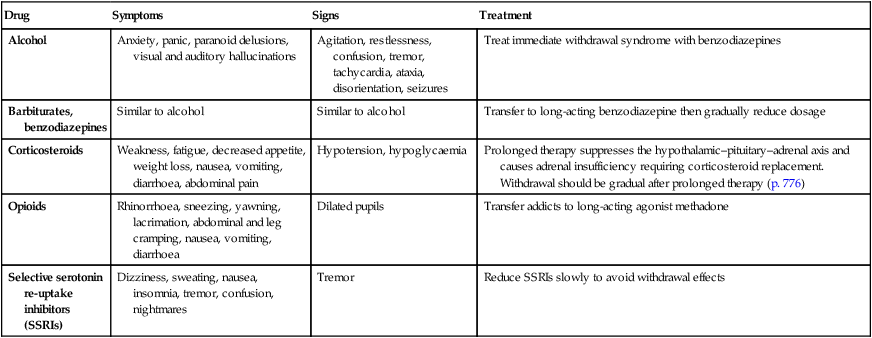
Desensitisation and withdrawal effects
 2.3 Examples of drugs associated with withdrawal effects
2.3 Examples of drugs associated with withdrawal effects
Drug
Symptoms
Signs
Treatment
Alcohol
Anxiety, panic, paranoid delusions, visual and auditory hallucinations
Agitation, restlessness, confusion, tremor, tachycardia, ataxia, disorientation, seizures
Treat immediate withdrawal syndrome with benzodiazepines
Barbiturates, benzodiazepines
Similar to alcohol
Similar to alcohol
Transfer to long-acting benzodiazepine then gradually reduce dosage
Corticosteroids
Weakness, fatigue, decreased appetite, weight loss, nausea, vomiting, diarrhoea, abdominal pain
Hypotension, hypoglycaemia
Prolonged therapy suppresses the hypothalamic–pituitary–adrenal axis and causes adrenal insufficiency requiring corticosteroid replacement. Withdrawal should be gradual after prolonged therapy (p. 776)
Opioids
Rhinorrhoea, sneezing, yawning, lacrimation, abdominal and leg cramping, nausea, vomiting, diarrhoea
Dilated pupils
Transfer addicts to long-acting agonist methadone
Selective serotonin re-uptake inhibitors (SSRIs)
Dizziness, sweating, nausea, insomnia, tremor, confusion, nightmares
Tremor
Reduce SSRIs slowly to avoid withdrawal effects
Pharmacokinetics
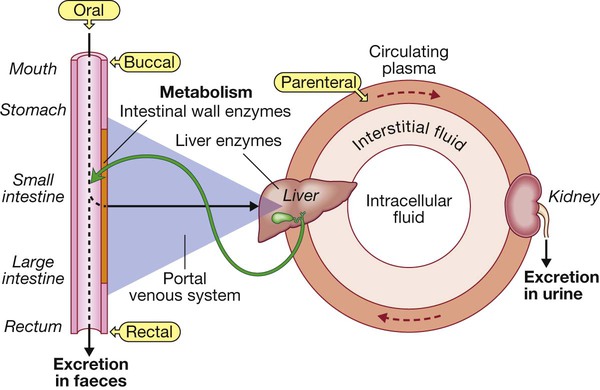
Most drugs are taken orally, are absorbed from the intestinal lumen and enter the portal venous system to be conveyed to the liver, where they may be subject to first-pass metabolism and/or excretion in bile. Active drugs then enter the systemic circulation, from which they may diffuse (or sometimes be actively transported) in and out of the interstitial and intracellular fluid compartments. Drug that remains in circulating plasma is subject to liver metabolism and renal excretion. Drugs excreted in bile may be reabsorbed, creating an enterohepatic circulation. First-pass metabolism in liver is avoided if drugs are administered via the buccal or rectal mucosa, or parenterally (e.g. by intravenous injection).
Drug absorption and routes of administration
Enteral administration
Parenteral administration
Other routes of administration
Drug distribution
Volume of distribution
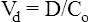
A In this example of first-order kinetics following a single intravenous dose, the time period required for the plasma drug concentration to halve (half-life, t1/2) remains constant throughout the elimination process. B After multiple dosing, the plasma drug concentration rises if each dose is administered before the previous dose has been entirely cleared. In this example, the drug’s half-life is 30 hours, so that with daily dosing the peak, average and trough concentrations steadily increase as drug accumulates in the body (black line). Steady state is reached after approximately 5 half-lives, when the rate of elimination (the product of concentration and clearance) is equal to the rate of drug absorption (the product of rate of administration and bioavailability). The long half-life in this example means that it takes 6 days for steady state to be achieved and, for most of the first 3 days of treatment, plasma drug concentrations are below the therapeutic range (yellow-shaded area). This problem can be overcome if a larger loading dose (red line) is used to achieve steady state drug concentrations more rapidly.
Drug elimination
Drug metabolism
Drug excretion
Elimination kinetics
Repeated dose regimens
Inter-individual variation in drug responses
Adverse outcomes of drug therapy
Adverse drug reactions
Prevalence of ADRs
 2.6 How to take a drug history
2.6 How to take a drug history
 2.7 Risk factors for ADRs
2.7 Risk factors for ADRs

 2.2
2.2
 2.4
2.4 2.5
2.5