384 | The Spondyloarthritides |
The spondyloarthritides are a group of overlapping disorders that share certain clinical features and genetic associations. These disorders include ankylosing spondylitis (AS), reactive arthritis, psoriatic arthritis and spondylitis, enteropathic arthritis and spondylitis, juvenile-onset spondyloarthritis (SpA), and undifferentiated SpA. The similarities in clinical manifestations and genetic predisposition suggest that these disorders share pathogenic mechanisms.
ANKYLOSING SPONDYLITIS
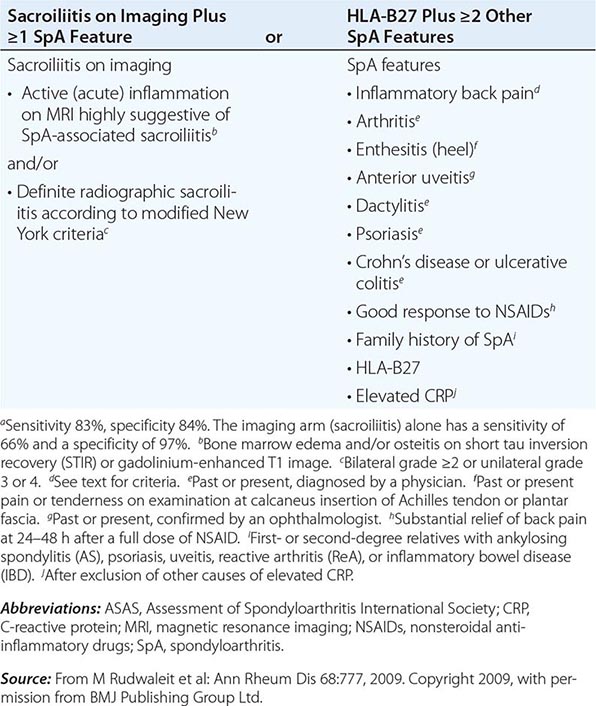
AS is an inflammatory disorder of unknown cause that primarily affects the axial skeleton; peripheral joints and extraarticular structures are also frequently involved. The disease usually begins in the second or third decade; male-to-female prevalence is between 2:1 and 3:1. The term axial spondyloarthritis is coming into common use, supported by criteria formulated in 2009 (Table 384-1). This classification includes both definite AS and early stages that do not yet meet classical criteria for AS, but it probably also includes other conditions with a different natural history.
ASAS CRITERIA FOR CLASSIFICATION OF AXIAL SPONDYLOARTHRITIS (TO BE APPLIED FOR PATIENTS WITH BACK PAIN ≥3 MONTHS AND AGE OF ONSET <45 YEARS)a |
EPIDEMIOLOGY
AS shows a striking correlation with the histocompatibility antigen HLA-B27 and occurs worldwide roughly in proportion to the prevalence of B27 (Chap. 373e). In North American whites, the prevalence of B27 is 7%, whereas it is 90% in patients with AS, independent of disease severity.
In population surveys, AS is present in 1–6% of adults inheriting B27, whereas the prevalence is 10–30% among B27+ adult first-degree relatives of AS probands. Concordance rate in identical twins is about 65%. Susceptibility to AS is determined largely by genetic factors, with B27 comprising less than one-half of the genetic component. Genomewide single-nucleotide polymorphism (SNP) analysis has identified over 30 additional susceptibility alleles.
PATHOLOGY
Sacroiliitis is often the earliest manifestation of AS. Knowledge of its pathology comes from both biopsy and autopsy studies that cover a range of disease durations. Synovitis and myxoid marrow represent the earliest changes, followed by pannus and subchondral granulation tissue. Marrow edema, enthesitis, and chondroid differentiation are also found. Macrophages, T cells, plasma cells, and osteoclasts are prevalent. Eventually the eroded joint margins are gradually replaced by fibrocartilage regeneration and then by ossification. The joint may become totally obliterated.
In the spine, the specimens studied have either been surgically resected in advanced disease or taken from autopsies. There is inflammatory granulation tissue in the paravertebral connective tissue at the junction of annulus fibrosus and vertebral bone, and in some cases along the entire outer annulus. The outer annular fibers are eroded and eventually replaced by bone, forming the beginning of a syndesmophyte, which then grows by continued endochondral ossification, ultimately bridging the adjacent vertebral bodies. Ascending progression of this process leads to the “bamboo spine.” Other lesions in the spine include diffuse osteoporosis (loss of trabecular bone despite accretion of periosteal bone), erosion of vertebral bodies at the disk margin, “squaring” or “barreling” of vertebrae, and inflammation and destruction of the disk-bone border. Inflammatory arthritis of the apophyseal (facet) joints is common, with synovitis, inflammation at the bony attachment of the joint capsule, and subchondral bone marrow granulation tissue. Erosion of joint cartilage by pannus is often followed by bony ankylosis. This may precede formation of syndesmophytes bridging the adjacent disks. Bone mineral density is diminished in the spine and proximal femur early in the course of the disease.
Peripheral synovitis in AS shows marked vascularity, which is also evident as tortuous macrovascularity seen during arthroscopy. Lining layer hyperplasia, lymphoid infiltration, and pannus formation are also found. Central cartilaginous erosions caused by proliferation of subchondral granulation tissue are common. It should be emphasized that the characteristics of peripheral arthritis in AS and other forms of SpA are similar, and distinct from those of rheumatoid arthritis.
Inflammation in the fibrocartilaginous enthesis, the region where a tendon, ligament, or joint capsule attaches to bone, is a characteristic lesion in AS and other SpAs, both at axial and peripheral sites. Enthesitis is associated with prominent edema of the adjacent bone marrow and is often characterized by erosive lesions that eventually undergo ossification.
Subclinical intestinal inflammation has been found in the colon or distal ileum in a majority of patients with SpA. The histology is described below under “Enteropathic Arthritis.”
PATHOGENESIS
The pathogenesis of AS is immune-mediated, but there is little direct evidence for antigen-specific autoimmunity, and there is evidence to suggest more of an autoinflammatory pathogenesis. Uncertainty remains regarding the primary site of disease initiation. The dramatic response of the disease to therapeutic blockade of tumor necrosis factor α (TNF-α) indicates that this cytokine plays a central role in the immunopathogenesis of AS. Other genes related to TNF pathways show association with AS, including TNFRSF1A, LTBR, and TBKBP1. More recent evidence strongly implicates the interleukin (IL) 23/IL-17 cytokine pathway in AS pathogenesis. At least five genes in this pathway show association with AS, including IL23R, PTER4, IL12B, CARD9, and TYK2. All of these genes are also associated with inflammatory bowel disease (IBD), and three of them are associated with psoriasis. Serum levels of IL-23 and IL-17 are elevated in AS patients. Mice expressing high levels of IL-23 show spontaneous infiltration in the entheses of CD3+CD4–CD8– cells bearing IL-23 receptors and producing IL-17 and IL-22. This finding suggests the possibility that site-specific innate immune cells may play a critical role in the anatomic specificity of the lesions. Mast cells and, to a lesser extent, neutrophils appear to be the major IL-17-producing cells in peripheral arthritis, whereas neutrophils producing IL-17 are prominent in apophyseal joints. High levels of circulating γδ T cells expressing IL-23 receptors and producing IL-17 have been found in AS patients.
Other associated genes encode other cytokines or cytokine receptors (IL6R, IL1R1, IL1R2, IL7R, IL27), transcription factors involved in the differentiation of immune cells (RUNX3, EOMES, BACH2, NKX2-3, TBX21), or other molecules involved in activation or regulation of immune or inflammatory responses (FCGR2A, ZMIZ1, NOS2, ICOSLG).
The inflamed sacroiliac joint is infiltrated with CD4+ and CD8+ T cells and macrophages and shows high levels of TNF-α, particularly early in the disease. Abundant transforming growth factor β (TGF-β) has been found in more advanced lesions. Peripheral synovitis in AS and the other spondyloarthritides is characterized by neutrophils, macrophages expressing CD68 and CD163, CD4+ and CD8+ T cells, and B cells. There is prominent staining for intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), matrix metalloproteinase 3 (MMP-3), and myeloid-related proteins 8 and 14 (MRP-8 and MRP-14). Unlike rheumatoid arthritis (RA) synovium, citrullinated proteins and cartilage gp39 peptide–major histocompatibility complexes (MHCs) are absent. However, citrullinated proteins can be seen in the circulation.
No specific event or exogenous agent that triggers the onset of disease has been identified, although overlapping features with reactive arthritis and IBD and the involvement of the IL-23/IL-17 pathway suggest that enteric bacteria may play a role, and microdamage from mechanical stress at enthesial sites has also been implicated.
It is firmly established that HLA-B27 plays a direct role in AS pathogenesis, but its precise role at the molecular level remains unresolved. Rats transgenic for HLA-B27 develop arthritis and spondylitis, and this is un-affected by the absence of CD8. It thus appears that classical peptide antigen presentation to CD8+ T cells may not be the primary disease mechanism. However, the association of AS with ERAP1, which strongly influences the MHC class I peptide repertoire, is only found in B27+ patients, and this suggests that peptide binding to B27 is nonetheless important. The pairs of ERAP1 alleles found in AS patients show diminished peptidase activity, compared with those found in healthy controls. The B27 heavy chain has an unusual tendency to misfold, a process that may be proinflammatory. Genetic and functional studies in humans have suggested a role for natural killer (NK) cells in AS, possibly through interaction with B27 heavy chain homodimers. SpA-prone B27 rats show defective dendritic cell function and share with AS patients a characteristic “reverse interferon” gene expression signature in antigen-presenting cells.
New bone formation in AS appears to be largely based on enchondral bone formation and occurs only in the periosteal compartment. It correlates with lack of regulation of the Wnt signaling pathway, which controls the differentiation of mesenchymal cells into osteophytes, by the inhibitors DKK-1 and sclerostin. Indirect evidence and data from animal models also implicate bone morphogenic proteins, hedgehog proteins, and prostaglandin E2. There is sharp controversy as to whether vertebral new bone formation in AS is a sequela of inflammation or whether it arises independently of inflammation. The second hypothesis is based on the observation that syndesmophyte formation is not suppressed by anti-TNF-α therapy that potently suppresses inflammation. TNF-α is also a known inducer of DKK-1, which inhibits bone formation. Recent magnetic resonance imaging (MRI) studies suggest that it is vertebral inflammatory lesions that undergo metaplasia to fat (increased T1-weighted signal) that are the predominant site of subsequent syndesmophytes despite anti-TNF-α therapy, whereas early acute inflammatory lesions resolve. A recent study suggested that the rate of syndesmophyte formation decreases after >4 years of anti-TNF-α therapy.
CLINICAL MANIFESTATIONS
The symptoms of the disease are usually first noticed in late adolescence or early adulthood; the median age in Western countries is approximately 23 years. In 5% of patients, symptoms begin after age 40. The initial symptom is usually dull pain, insidious in onset, felt deep in the lower lumbar or gluteal region, accompanied by low-back morning stiffness of up to a few hours’ duration that improves with activity and returns following inactivity. Within a few months, the pain has usually become persistent and bilateral. Nocturnal exacerbation of pain often forces the patient to rise and move around.
In some patients, bony tenderness (presumably reflecting enthesitis or osteitis) may accompany back pain or stiffness, whereas in others it may be the predominant complaint. Common sites include the costosternal junctions, spinous processes, iliac crests, greater trochanters, ischial tuberosities, tibial tubercles, and heels. Hip and shoulder (“root” joint) arthritis is considered part of the axial disease. Hip arthritis occurs in 25–35% of patients. Shoulder arthritis is much less common. Severe isolated hip arthritis or bony chest pain may be the presenting complaint, and symptomatic hip disease can dominate the clinical picture. Arthritis of peripheral joints other than the hips and shoulders, usually asymmetric, occurs in up to 30% of patients. Neck pain and stiffness from involvement of the cervical spine are usually relatively late manifestations but are occasionally dominant symptoms. Rare patients, particularly in the older age group, present with predominantly constitutional symptoms.
AS often has a juvenile onset in developing countries. Peripheral arthritis and enthesitis usually predominate, with axial symptoms supervening in late adolescence.
Initially, physical findings mirror the inflammatory process. The most specific findings involve loss of spinal mobility, with limitation of anterior and lateral flexion and extension of the lumbar spine and of chest expansion. Limitation of motion is usually out of proportion to the degree of bony ankylosis and is thought to possibly reflect muscle spasm secondary to pain and inflammation. Pain in the sacroiliac joints may be elicited either with direct pressure or with stress on the joints. In addition, there is commonly tenderness upon palpation of the posterior spinous processes and other sites of symptomatic bony tenderness.
The modified Schober test is a useful measure of lumbar spine flexion. The patient stands erect, with heels together, and marks are made on the spine at the lumbosacral junction (identified by a horizontal line between the posterosuperior iliac spines) and 10 cm above. The patient then bends forward maximally with knees fully extended, and the distance between the two marks is measured. This distance increases by ≥5 cm in the case of normal mobility and by <4 cm in the case of decreased mobility. Chest expansion is measured as the difference between maximal inspiration and maximal forced expiration in the fourth intercostal space in males or just below the breasts in females, with the patient’s hands resting on or just behind the head. Normal chest expansion is ≥5 cm. Lateral bending measures the distance the patient’s middle finger travels down the leg with maximal lateral bending. Normal is >10 cm.
Limitation or pain with motion of the hips or shoulders is usually present if these joints are involved. It should be emphasized that early in the course of mild cases, symptoms may be subtle and nonspecific, and the physical examination may be unrevealing.
The course of the disease is extremely variable, ranging from the individual with mild stiffness and normal radiographs to the patient with a totally fused spine and severe bilateral hip arthritis, accompanied by severe peripheral arthritis and extraarticular manifestations. Pain tends to be persistent early in the disease and intermittent later, with alternating exacerbations and quiescent periods. In a typical severe untreated case with progression of the spondylitis to syndesmophyte formation, the patient’s posture undergoes characteristic changes, with obliterated lumbar lordosis, buttock atrophy, and accentuated thoracic kyphosis. There may be a forward stoop of the neck or flexion contractures at the hips, compensated by flexion at the knees. Disease progression can be estimated clinically from loss of height, limitation of chest expansion and spinal flexion, and occiput-to-wall distance. Occasional individuals are encountered with advanced deformities who report having never had significant symptoms.
The factors most predictive of radiographic progression (see below) are the presence of existing syndesmophytes, high inflammatory makers, and smoking. In some but not all studies, onset of AS in adolescence and early hip involvement correlate with a worse prognosis. In women, AS tends to progress less frequently to total spinal ankylosis, although there may be an increased prevalence of isolated cervical ankylosis and peripheral arthritis. In industrialized countries, peripheral arthritis (distal to hips and shoulders) occurs in less than one-half of patients with AS, usually as a late manifestation, whereas in developing countries, the prevalence is much higher, with onset typically early in the disease course. Pregnancy has no consistent effect on AS, with symptoms improving, remaining the same, or deteriorating in one-third of pregnant patients, respectively.
The most serious complication of the spinal disease is spinal fracture, which can occur with even minor trauma to the rigid, osteoporotic spine. The lower cervical spine is most commonly involved. These fractures are often displaced and cause spinal cord injury. A recent survey suggested a >10% lifetime risk of fracture. Occasionally, fracture through a diskovertebral junction and adjacent neural arch, termed pseudoarthrosis, most common in the thoracolumbar spine, can be an unrecognized source of persistent localized pain and/or neurologic dysfunction. Wedging of thoracic vertebrae is common and correlates with accentuated kyphosis.
The most common extraarticular manifestation is acute anterior uveitis, which occurs in up to 40% of patients and can antedate the spondylitis. Attacks are typically unilateral, causing pain, photophobia, and increased lacrimation. These tend to recur, often in the opposite eye. Cataracts and secondary glaucoma are not uncommon sequelae. Up to 60% of patients with AS have inflammation in the colon or ileum. This is usually asymptomatic, but frank IBD occurs in 5–10% of patients with AS (see “Enteropathic Arthritis,” below). About 10% of patients meeting criteria for AS have psoriasis (see “Psoriatic Arthritis,” below). Aortic insufficiency, sometimes leading to congestive heart failure, occurs in a small percentage of patients, occasionally early. Third-degree heart block may occur alone or together with aortic insufficiency. Subclinical pulmonary lesions and cardiac dysfunction may be relatively common. Cauda equina syndrome and upper pulmonary lobe fibrosis are rare late complications. Retroperitoneal fibrosis is a rare associated condition. Prostatitis has been reported to have an increased prevalence. Amyloidosis is rare (Chap. 137).
Several validated measures of disease activity and functional outcome are in widespread use in the study and management of AS, particularly the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and the Ankylosing Spondylitis Disease Activity Score (ASDAS), both measures of disease activity; the Bath Ankylosing Spondylitis Functional Index (BASFI), a measure of limitation in activities of daily living; and several measures of radiographic changes. The Harris hip score, although not specific for AS, can be helpful. Despite persistence of the disease, most patients remain gainfully employed. Some but not all studies of survival in AS have suggested that AS shortens life span, compared with the general population. Mortality attributable to AS is largely the result of spinal trauma, aortic insufficiency, respiratory failure, amyloid nephropathy, or complications of therapy such as upper gastrointestinal hemorrhage. The impact of anti-TNF therapy on outcome and mortality is not yet known, except for significantly improved work productivity.
LABORATORY FINDINGS
No laboratory test is diagnostic of AS. In most ethnic groups, HLA-B27 is present in 80–90% of patients. Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are often, but not always, elevated. Mild anemia may be present. Patients with severe disease may show an elevated alkaline phosphatase level. Elevated serum IgA levels are common. Rheumatoid factor, anti-cyclic citrullinated peptide (CCP), and antinuclear antibodies (ANAs) are largely absent unless caused by a coexistent disease, although ANAs may appear with anti-TNF therapy. Circulating levels of CD8+ T cells tend to be low, and serum matrix metalloproteinase 3 levels correlate with disease activity. Synovial fluid from peripheral joints in AS is nonspecifically inflammatory. In cases with restriction of chest wall motion, decreased vital capacity and increased functional residual capacity are common, but airflow is normal and ventilatory function is usually well maintained.
RADIOGRAPHIC FINDINGS
Radiographically demonstrable sacroiliitis, usually symmetric, is eventually present in AS. The earliest changes by standard radiography are blurring of the cortical margins of the subchondral bone, followed by erosions and sclerosis. Progression of the erosions leads to “pseudowidening” of the joint space; as fibrous and then bony ankylosis supervene, the joints may become obliterated.
In the lumbar spine, progression of the disease leads to straightening, caused by loss of lordosis, and reactive sclerosis, caused by osteitis of the anterior corners of the vertebral bodies with subsequent erosion, leading to “squaring” or even “barreling” of one or more vertebral bodies. Progressive ossification leads to eventual formation of marginal syndesmophytes, visible on plain films as bony bridges connecting successive vertebral bodies anteriorly and laterally.
Years may elapse before unequivocal sacroiliac abnormalities are evident on plain radiographs, and consequently, MRI is being increasingly used in diagnosing AS. Active sacroiliitis is best visualized by dynamic MRI with fat saturation, either T2-weighed turbo spin-echo sequence or short tau inversion recovery (STIR) with high resolution, or T1-weighted images with contrast enhancement. These techniques sensitively identify early intraarticular inflammation, cartilage changes, and underlying bone marrow edema in sacroiliitis (Fig. 384-1). They are also highly sensitive for evaluation of acute and chronic spinal changes (Fig. 384-2).
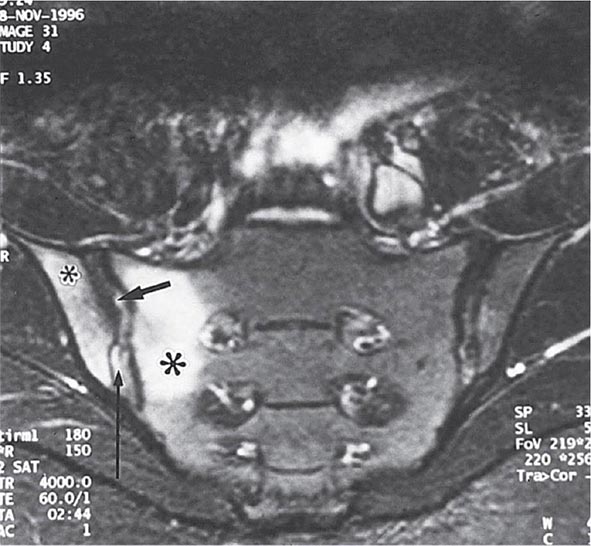
FIGURE 384-1 Early sacroiliitis in a patient with ankylosing spondylitis, indicated by prominent edema in the juxtaarticular bone marrow (asterisks), synovium and joint capsule (thin arrow), and interosseous ligaments (thick arrow) on a short tau inversion recovery (STIR) magnetic resonance image. (From M Bollow et al: Zeitschrift für Rheumatologie 58:61, 1999. Reproduced with permission.)
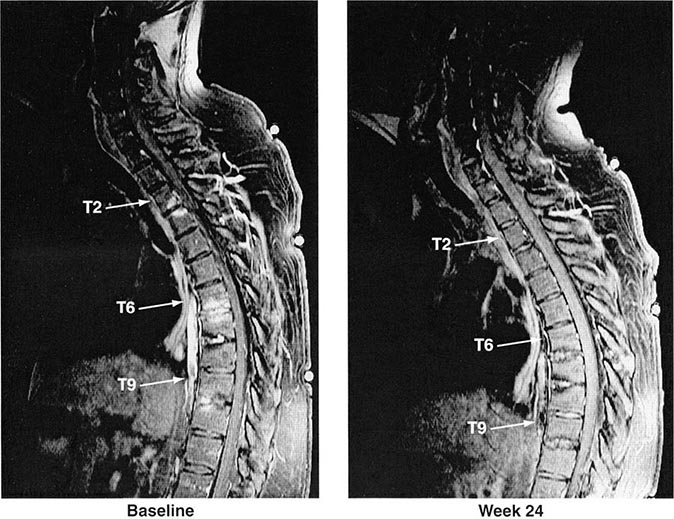
FIGURE 384-2 Spinal inflammation (spondylodiskitis) in a patient with ankylosing spondylitis and its dramatic response to treatment with infliximab. Gadolinium-enhanced T1-weighted magnetic resonance images, with fat saturation, at baseline and after 24 weeks of infliximab therapy. (From J Braun et al: Arthritis Rheum 54:1646, 2006.)
Reduced bone mineral density can be detected by dual-energy x-ray absorptiometry of the femoral neck and the lumbar spine. Use of a lateral projection of the L3 vertebral body can prevent falsely elevated readings related to spinal ossification.
DIAGNOSIS
It is important to establish the diagnosis of early AS before the development of irreversible deformity. This goal presents a challenge for several reasons: (1) Back pain is very common, but AS is much less common; (2) an early presumptive diagnosis often relies on clinical grounds requiring considerable expertise; and (3) young individuals with symptoms of AS often do not seek medical care. The widely used modified New York criteria (1984) are based on the presence of definite radiographic sacroiliitis and are too insensitive in early or mild cases. In 2009, new criteria for axial SpA were proposed by the Assessment of Spondyloarthritis International Society (ASAS) (Table 384-1). They are applicable to individuals with ≥3 months of back pain with age of onset <45 years old. Active inflammation of the sacroiliac joints as determined by dynamic MRI is considered equivalent to definite radiographic sacroiliitis (see below).
AS must be differentiated from numerous other causes of low-back pain, some far more common than AS. To qualify as the criterion for inflammatory back pain of axial SpA (Table 384-1), the chronic (≥3 months) back pain should have four or more of the following characteristic features: (1) age of onset <40 years old; (2) insidious onset; (3) improvement with exercise; (4) no improvement with rest; and (5) pain at night with improvement upon getting up. Other common features of inflammatory back pain include morning stiffness >30 min, awakening from back pain during only the second half of the night, and alternating buttock pain. In clinical decision-making, all of these features are additive. The most common causes of back pain other than AS are primarily mechanical or degenerative rather than primarily inflammatory and tend not to show clustering of these features.
Less-common metabolic, infectious, and malignant causes of back pain must also be differentiated from AS, including infectious spondylitis, spondylodiskitis, and sacroiliitis, and primary or metastatic tumor. Ochronosis can produce a phenotype that is clinically and radiographically similar to AS. Calcification and ossification of paraspinous ligaments occur in diffuse idiopathic skeletal hyperostosis (DISH), which occurs in the middle-aged and elderly and is usually not symptomatic. Ligamentous calcification gives the appearance of “flowing wax” on the anterior bodies of the vertebrae. Intervertebral disk spaces are preserved, and sacroiliac and apophyseal joints appear normal, helping to differentiate DISH from spondylosis and from AS, respectively.
REACTIVE ARTHRITIS
Reactive arthritis (ReA) refers to acute nonpurulent arthritis complicating an infection elsewhere in the body. In recent years, the term has been used primarily to refer to SpA following enteric or urogenital infections.
Other forms of reactive and infection-related arthritis not associated with B27 and showing a spectrum of clinical features different from SpA, such as Lyme disease and rheumatic fever, are discussed in Chaps. 210 and 381.
HISTORIC BACKGROUND
The association of acute arthritis with episodes of diarrhea or urethritis has been recognized for centuries. A large number of cases during World Wars I and II focused attention on the triad of arthritis, urethritis, and conjunctivitis, often with additional mucocutaneous lesions, which became widely known by eponyms that are now of historic interest only.
The identification of bacterial species capable of triggering the clinical syndrome and the finding that many patients possess the B27 antigen led to the unifying concept of ReA as a clinical syndrome triggered by specific etiologic agents in a genetically susceptible host. A similar spectrum of clinical manifestations can be triggered by enteric infection with any of several Shigella, Salmonella, Yersinia, and Campylobacter species; by genital infection with Chlamydia trachomatis; and by other agents as well. The triad of arthritis, urethritis, and conjunctivitis represents a small part of the spectrum of the clinical manifestations of ReA and only a minority of patients present with this “classic triad” of symptoms. Although emerging data suggest that asymptomatic Chlamydia trachomatis infections might trigger ReA, for the purposes of this chapter, the use of the term ReA will be restricted to those cases of SpA in which there is at least presumptive evidence for a related symptomatic antecedent infection. Patients with clinical features of ReA who lack evidence of an antecedent infection will be considered to have undifferentiated spondyloarthritis, discussed below.
EPIDEMIOLOGY
Initial reports may have overestimated the association of ReA with HLA-B27, since 60–85% of patients who developed ReA triggered by Shigella, Yersinia, or Chlamydia were B27-positive. However, other studies demonstrated a lower prevalence of B27 in ReA triggered by Salmonella, with one study suggesting no association in Campylobacter-induced ReA. Several more recent community-based or common-source epidemic studies demonstrated that the prevalence of B27 in ReA was below 50%. The most common age range is 18–40 years, but ReA can occur rarely in children and occasionally in older adults.
The attack rate of postenteric ReA generally ranges from 1% to about 30% depending on the study and causative organism, whereas the attack rate of postchlamydial ReA is about 4–8%. The gender ratio in ReA following enteric infection is nearly 1:1, whereas venereally acquired ReA occurs mainly in men. The overall prevalence and incidence of ReA are difficult to assess because of the lack of validated diagnostic criteria, variable prevalence and arthritogenic potential of the triggering infectious agents, and inconstant genetic susceptibility factors in different populations. In Scandinavia, an annual incidence of 10–28:100,000 has been reported. The spondyloarthritides were formerly almost unknown in sub-Saharan Africa. However, ReA and other peripheral SpAs have now become the most common rheumatic diseases in Africans in the wake of the AIDS epidemic, without association to B27, which is very rare in these populations. ReA is often the first manifestation of HIV infection and often remits with disease progression. In contrast, Western white patients with HIV and SpA are usually B27-positive, and the arthritis flares as AIDS advances.
PATHOLOGY
Synovial histology is similar to that of other SpAs. Enthesitis shows increased vascularity and macrophage infiltration of fibrocartilage. Microscopic histopathologic evidence of inflammation mimicking IBD has routinely been demonstrated in the colon and ileum of patients with postenteric ReA and less commonly in postvenereal ReA. The skin lesions of keratoderma blennorrhagica, associated mainly with venereally acquired ReA, are histologically indistinguishable from pustular psoriatic lesions.
ETIOLOGY AND PATHOGENESIS
The bacteria identified as definitive triggers of ReA include several Salmonella spp., Shigella spp., Yersinia enterocolitica, Yersinia pseudotuberculosis, Campylobacter jejuni, and Chlamydia trachomatis. These triggering microbes are gram-negative bacteria with a lipopolysaccharide component to their cell walls. All four Shigella species (Shigella sonnei, Shigella boydii, Shigella flexneri, and Shigella dysenteriae) have been implicated in cases of ReA, with S. flexneri and S. sonnei being the most common. After Salmonella infection, individuals of Caucasian descent may be more likely than those of Asian descent to develop ReA. Children may be less susceptible to ReA caused by Salmonella and Campylobacter. Yersinia species in Europe and Scandinavia may have greater arthritogenic potential than in other parts of the world, and C. trachomatis appears to be a common cause worldwide. The ocular serovars of C. trachomatis appear to be particularly, perhaps uniquely, arthritogenic.
There is also evidence implicating Clostridium difficile, Campylobacter coli, certain toxigenic Escherichia coli, and possibly Ureaplasma urealyticum and Mycoplasma genitalium as potential triggers of ReA. Chlamydia pneumoniae is another trigger of ReA, albeit far less common than C. trachomatis. There have also been numerous isolated reports of acute arthritis preceded by other bacterial, viral, or parasitic infections, and even following intravesicular bacillus Calmette-Guérin (BCG) treatment for bladder cancer.
It has not been determined whether ReA occurs by the same pathogenic mechanism following infection with each of these microorganisms, nor has the mechanism been elucidated in the case of any one of the known bacterial triggers. Most, if not all, of the organisms well established to be triggers share a capacity to attack mucosal surfaces, to invade host cells, and to survive intracellularly. Antigens from Chlamydia, Yersinia, Salmonella, and Shigella have been shown to be present in the synovium and/or synovial fluid leukocytes of patients with ReA for long periods following the acute attack. In ReA triggered by Y. enterocolitica, bacterial lipopolysaccharide (LPS) and heat-shock protein antigens have been found in peripheral blood cells years after the triggering infection. Yersinia DNA and C. trachomatis DNA and RNA have been detected in synovial tissue from ReA patients, suggesting the presence of viable organisms despite uniform failure to culture the organism from these specimens. In C. trachomatis–induced ReA specifically, the bacterial load in synovial tissue of patients with remitting disease is lower than that of active disease, but mRNAs encoding proinflammatory proteins are equal to or higher than those of active disease. The specificity of these findings is unclear, however, since chromosomal bacterial DNA and 16S rRNA from a very wide variety of bacteria have also been found in synovium in other rheumatic diseases, albeit less frequently. In several older studies, synovial T cells that specifically responded to antigens of the inciting organism were reported and characterized as predominantly CD4+ with a TH2 or T regulatory phenotype. More recent work has documented high levels of IL-17 in ReA synovial fluid, but the source has not been identified. HLA-B27 seems to be associated with more severe and chronic forms of the “classic triad” of ReA, but its pathogenic role remains to be determined. HLA-B27 significantly prolongs the intracellular survival of Y. enterocolitica and Salmonella enteritidis in human and mouse cell lines. Prolonged intracellular bacterial survival, promoted by B27, other factors, or both, may permit trafficking of infected leukocytes from the site of primary infection to joints, where an innate and/or adaptive immune response to persistent bacterial antigens may then promote arthritis.
CLINICAL FEATURES
The clinical manifestations of ReA constitute a spectrum that ranges from an isolated, transient monoarthritis or enthesitis to severe multisystem disease. A careful history will usually elicit evidence of an antecedent infection 1–4 weeks before onset of symptoms of the reactive disease, particularly in postenteric ReA. However, in a sizable minority, no clinical or laboratory evidence of an antecedent infection can be found, particularly in the case of postchlamydial ReA. In cases of presumed venereally acquired reactive disease, there is often a history of a recent new sexual partner, even without laboratory evidence of infection.
Constitutional symptoms are common, including fatigue, malaise, fever, and weight loss. The musculoskeletal symptoms are usually acute in onset. Arthritis is usually asymmetric and additive, with involvement of new joints occurring over a few days to 1–2 weeks. The joints of the lower extremities, especially the knee, ankle, and subtalar, metatarsophalangeal, and toe interphalangeal joints, are most commonly involved, but the wrist and fingers can be involved as well. The arthritis is usually quite painful, and tense joint effusions are not uncommon, especially in the knee. Dactylitis, or “sausage digit,” a diffuse swelling of a solitary finger or toe, is a distinctive feature of ReA and other peripheral spondyloarthritides but can be seen in polyarticular gout and sarcoidosis. Tendinitis and fasciitis are particularly characteristic lesions, producing pain at multiple insertion sites (entheses), especially the Achilles insertion, the plantar fascia, and sites along the axial skeleton. Spinal, low-back, and buttock pain are quite common and may be caused by insertional inflammation, muscle spasm, acute sacroiliitis, or, presumably, arthritis in intervertebral joints.
Urogenital lesions may occur throughout the course of the disease. In males, urethritis may be marked or relatively asymptomatic and may be either an accompaniment of the triggering infection or a result of the reactive phase of the disease; interestingly, it occurs in both postvenereal and postenteric ReA. Prostatitis is also common. Similarly, in females, cervicitis or salpingitis may be caused either by the infectious trigger or by the sterile reactive process.
Ocular disease is common, ranging from transient, asymptomatic conjunctivitis to an aggressive anterior uveitis that occasionally proves refractory to treatment and may result in blindness.
Mucocutaneous lesions are frequent. Oral ulcers tend to be superficial, transient, and often asymptomatic. The characteristic skin lesions, keratoderma blennorrhagica, consist of vesicles and/or pustules that become hyperkeratotic, ultimately forming a crust before disappearing. They are most common on the palms and soles but may occur elsewhere as well. In patients with HIV infection, these lesions are often extremely severe and extensive, sometimes dominating the clinical picture (Chap. 226). Lesions may occur on the glans penis, termed circinate balanitis; these consist of vesicles that quickly rupture to form painless superficial erosions, which in circumcised individuals can form crusts similar to those of keratoderma blennorrhagica. Nail changes are common and consist of onycholysis, distal yellowish discoloration, and/or heaped-up hyperkeratosis.
Less-frequent or rare manifestations of ReA include cardiac conduction defects, aortic insufficiency, central or peripheral nervous system lesions, and pleuropulmonary infiltrates.
Arthritis typically persists for 3–5 months, but more chronic courses do occur. Chronic joint symptoms persist in about 15% of patients and in up to 60% of patients in hospital-based series, but these tend to be less severe than in the acute stage. Recurrences of the acute syndrome are also common. Work disability or forced change in occupation is common in those with persistent joint symptoms. Chronic heel pain is often particularly distressing. Low-back pain, sacroiliitis, and frank AS are also common sequelae. In most studies, HLA-B27–positive patients have shown a worse outcome than B27-negative patients. Patients with Yersinia– or Salmonella-induced arthritis have less chronic disease than those whose initial episode follows epidemic shigellosis.
LABORATORY AND RADIOGRAPHIC FINDINGS
The ESR and acute-phase reactants are usually elevated during the acute phase of the disease, often markedly so. Mild anemia may be present. Synovial fluid is nonspecifically inflammatory. In most ethnic groups, 30–50% of the patients are B27-positive. The triggering infection usually does not persist at the site of primary mucosal infection through the time of onset of the reactive disease, but it may be possible to culture the organism, e.g., in the case of Yersinia– or Chlamydia-induced disease. Serologic evidence of exposure to one of the causative organisms with elevation of antibodies is nonspecific and is of questionable utility. Polymerase chain reaction (PCR) for chlamydial DNA in first-voided urine specimens may have high sensitivity in the acute stage but is less useful with chronic disease.
In early or mild disease, radiographic changes may be absent or confined to juxtaarticular osteoporosis. With long-standing persistent disease, radiographic features share those of psoriatic arthritis; marginal erosions and loss of joint space can be seen in affected joints. Periostitis with reactive new bone formation is characteristic, as in all the SpAs. Spurs at the insertion of the plantar fascia are common.
Sacroiliitis and spondylitis may be seen as late sequelae. Sacroiliitis is more commonly asymmetric than in AS, and spondylitis, rather than ascending symmetrically, can begin anywhere along the lumbar spine. The syndesmophytes are described as nonmarginal; they are coarse, asymmetric, and “comma”-shaped, arising from the middle of a vertebral body, a pattern less commonly seen in primary AS. Progression to spinal fusion is uncommon.
DIAGNOSIS
ReA is a clinical diagnosis with no definitively diagnostic laboratory test or radiographic finding. The diagnosis should be entertained in any patient with an acute inflammatory, asymmetric, additive arthritis or tendinitis. The evaluation should include questioning regarding possible triggering events such as an episode of diarrhea or dysuria. On physical examination, attention must be paid to the distribution of the joint and tendon involvement and to possible sites of extraarticular involvement, such as the eyes, mucous membranes, skin, nails, and genitalia. Synovial fluid analysis may be helpful in excluding septic or crystal-induced arthritis. Culture, serology, or molecular methods may help to identify a triggering infection, but they cannot be relied upon.
Although typing for B27 has low negative predictive value in ReA, it may have prognostic significance in terms of severity, chronicity, and the propensity for spondylitis and uveitis. Furthermore, if positive, it can be helpful diagnostically in atypical cases. HIV testing is often indicated and may be necessary in order to select appropriate therapy.
It is important to differentiate ReA from disseminated gonococcal disease (Chap. 181), both of which can be venereally acquired and associated with urethritis. Unlike ReA, gonococcal arthritis and tenosynovitis tend to involve both upper and lower extremities equally, spare the axial skeleton, and be associated with characteristic vesicular skin lesions. A positive gonococcal culture from the urethra or cervix does not exclude a diagnosis of ReA; however, culturing gonococci from blood, skin lesion, or synovium establishes the diagnosis of disseminated gonococcal disease. PCR assay for Neisseria gonorrhoeae and C. trachomatis may be helpful. Occasionally, only a therapeutic trial of antibiotics can distinguish the two.
ReA shares many features in common with psoriatic arthropathy. However, psoriatic arthritis is usually gradual in onset; the arthritis tends to affect primarily the upper extremities; there is less associated periarthritis; and there are usually no associated mouth ulcers, urethritis, or bowel symptoms.
PSORIATIC ARTHRITIS
Psoriatic arthritis (PsA) refers to an inflammatory musculoskeletal disease that has both autoimmune and autoinflammatory features characteristically occurring in individuals with psoriasis.
HISTORIC BACKGROUND
The association between arthritis and psoriasis was noted in the nineteenth century. In the 1960s, on the basis of epidemiologic and clinical studies, it became clear that unlike RA, the arthritis associated with psoriasis was usually seronegative, often involved the distal interphalangeal (DIP) joints of the fingers and the spine and sacroiliac joints, had distinctive radiographic features, and showed considerable familial aggregation. In the 1970s, PsA was included in the broader category of the spondyloarthritides because of features similar to those of AS and ReA.
EPIDEMIOLOGY
Estimates of the prevalence of PsA among individuals with psoriasis range from 5 to 42%. The prevalence of PsA appears to be increasing in parallel with disease awareness; recent data using screening tools suggest that 20% or more of patients with psoriasis have undiagnosed PsA. The duration and severity of psoriasis increase one’s likelihood of developing PsA. In white populations, psoriasis is estimated to have a prevalence of 1–3%. Psoriasis and PsA are less common in other races in the absence of HIV infection, and the prevalence of PsA in individuals with psoriasis may be less common. First-degree relatives of PsA patients have an elevated risk for psoriasis, for PsA itself, and for other forms of SpA. Of patients with psoriasis, up to 30% have an affected first-degree relative. In monozygotic twins, the reported concordance for psoriasis varies from 35 to 72%, and for PsA from 10 to 30%. A variety of HLA associations have been found. The HLA-Cw*0602 gene is directly associated with psoriasis, particularly familial juvenile-onset (type I) psoriasis. HLA-B27 is associated with psoriatic spondylitis (see below). HLA-DR7, -DQ3, and -B57 are associated with PsA because of linkage disequilibrium with Cw6. Other associations with PsA include HLA-B13, -B37, -B38, -B39, -C12, and -DR4. A recent genome-wide scan found association of both psoriasis and PsA with a polymorphism at the HCP5 locus closely linked to HLA-B, and also to IL-23R, IL-12B (chromosome 5q31), IL-13, and several other chromosomal regions. Certain genetic loci are associated with PsA but not psoriasis, e.g., RUNX3 and IL-13.
PATHOLOGY
The inflamed synovium in PsA resembles that of RA, although with somewhat less hyperplasia and cellularity than in RA. As noted with AS above, the synovial vascular pattern in PsA is generally greater and more tortuous than in RA, independent of disease duration. Some studies have indicated a higher tendency to synovial fibrosis in PsA. Unlike RA, PsA shows prominent enthesitis, with histology similar to that of the other spondyloarthritides.
PATHOGENESIS
PsA is almost certainly immune-mediated and probably shares pathogenic mechanisms with psoriasis. PsA synovium is characterized by lining layer hyperplasia; diffuse infiltration with T cells, B cells, macrophages, and NK receptor–expressing cells, with upregulation of leukocyte homing receptors; and neutrophil proliferation with angiogenesis. Clonally expanded T cell subpopulations are frequent and have been demonstrated both in the synovium and the skin. Plasmacytoid dendritic cells are thought to play a key role in psoriasis, and there is some evidence for their participation in PsA. There is abundant synovial overexpression of proinflammatory cytokines, and synovial tissue staining has identified an overexpression of monocyte-derived cytokines, such as myeloid-related protein (S100A8/A9). Interferon γ, TNF-α, and IL-1β, -2, -6, -8, -10, -12, -13, and -15 are found in PsA synovium or synovial fluid. TH17-derived cytokines are important in PsA, given the genetic association with genes in the IL-12/IL-23 axis and the therapeutic response to an antibody to the shared IL-12/23 p40 subunit (see below). TH17 cells have been identified from the dermal extracts of psoriatic lesions and the synovial fluid of PsA patients. The majority of these CD4+ IL-17+ T cells are of memory phenotype (CD4RO[+]CD45RA[–]CD11a[+]). Consistent with the extensive bone remodeling in PsA, patients with PsA have been found to have a marked increase in osteoclastic precursors in peripheral blood and upregulation of receptor activator of nuclear factor κβ ligand (RANKL) in the synovial lining layer. Increased serum levels of TNF-α, RANKL, leptin, and omentin positively correlate with these osteoclastic precursors.
CLINICAL FEATURES
In 60–70% of cases, psoriasis precedes joint disease. In 15–20% of cases, the two manifestations appear within 1 year of each other. In about 15–20% of cases, the arthritis precedes the onset of psoriasis and can present a diagnostic challenge. The frequency in men and women is almost equal, although the frequency of disease patterns differs somewhat in the two sexes. The disease can begin in childhood or late in life but typically begins in the fourth or fifth decade, at an average age of 37 years.
The spectrum of arthropathy associated with psoriasis is quite broad. Many classification schemes have been proposed. In the original scheme of Wright and Moll, five patterns are described: (1) arthritis of the DIP joints; (2) asymmetric oligoarthritis; (3) symmetric polyarthritis similar to RA; (4) axial involvement (spine and sacroiliac joints); and (5) arthritis mutilans, a highly destructive form of disease. These patterns frequently coexist, and the pattern that persists chronically often differs from that of the initial presentation. A simpler scheme in recent use contains three patterns: oligoarthritis, polyarthritis, and axial arthritis.
Nail changes in the fingers or toes occur in up to 90% of patients with PsA, compared with 40% of psoriatic patients without arthritis, and pustular psoriasis is said to be associated with more severe arthritis. Several articular features distinguish PsA from other joint disorders; such hallmark features include dactylitis and enthesitis. Dactylitis occurs in >30%; enthesitis and tenosynovitis are also common and are probably present in most patients, although often not appreciated on physical examination. Shortening of digits because of underlying osteolysis is particularly characteristic of PsA (Fig. 384-3), and there is a much greater tendency than in RA for both fibrous and bony ankylosis of small joints. Rapid ankylosis of one or more proximal interphalangeal (PIP) joints early in the course of disease is not uncommon. Back and neck pain and stiffness are also common in PsA.
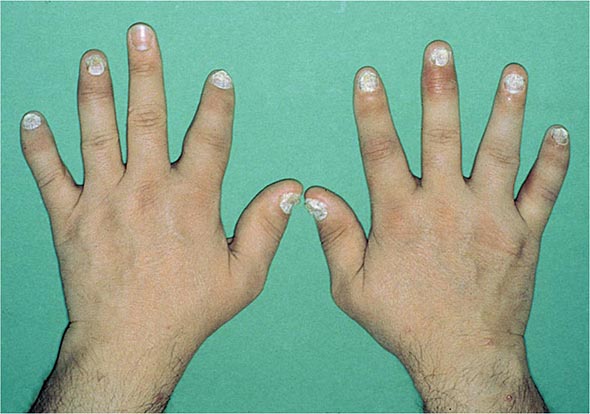
FIGURE 384-3 Characteristic lesions of psoriatic arthritis. Inflammation is prominent in the distal interphalangeal joints (left 5th, 4th, 2nd; right 2nd, 3rd, and 5th) and proximal interphalangeal joints (left 2nd, right 2nd, 4th, and 5th). There is dactylitis in the left 2nd finger and thumb, with pronounced telescoping of the left 2nd finger. Nail dystrophy (hyperkeratosis and onycholysis) affects each of the fingers except the left 3rd finger, the only finger without arthritis. (Courtesy of Donald Raddatz, MD; with permission.)
Arthropathy confined to the DIP joints occurs in about 5% of cases. Accompanying nail changes in the affected digits are almost always present. These joints are also often affected in the other patterns of PsA. Approximately 30% of patients have asymmetric oligoarthritis. This pattern commonly involves a knee or another large joint with a few small joints in the fingers or toes, often with dactylitis. Symmetric polyarthritis occurs in about 40% of PsA patients at presentation. It may be indistinguishable from RA in terms of the joints involved, but other features characteristic of PsA are usually also present. Almost any peripheral joint can be involved. Axial arthropathy without peripheral involvement is found in about 5% of PsA patients. It may be clinically indistinguishable from idiopathic AS, although more neck involvement and less thoracolumbar spinal involvement are characteristic, and nail changes are not found in idiopathic AS. A small percentage of PsA patients have arthritis mutilans, in which there can be widespread shortening of digits (“telescoping”), sometimes coexisting with ankylosis and contractures in other digits.
Six patterns of nail involvement are identified: pitting, horizontal ridging, onycholysis, yellowish discoloration of the nail margins, dystrophic hyperkeratosis, and combinations of these findings. Other extraarticular manifestations of the spondyloarthritides are common. Eye involvement, either conjunctivitis or uveitis, is reported in 7–33% of PsA patients. Unlike the uveitis associated with AS, the uveitis in PsA is more often bilateral, chronic, and/or posterior. Aortic valve insufficiency has been found in <4% of patients, usually after long-standing disease.
Widely varying estimates of clinical outcome have been reported in PsA. At its worst, severe PsA with arthritis mutilans is potentially at least as crippling and ultimately fatal as severe RA. Unlike RA, however, many patients with PsA experience temporary remissions. Overall, erosive disease develops in the majority of patients, progressive disease with deformity and disability is common, and in some large published series, mortality was found to be significantly increased compared with the general population. There appears to be a greater incidence of cardiovascular death in psoriatic disease.
The psoriasis and associated arthropathy seen with HIV infection both tend to be severe and can occur in populations with very little psoriasis in noninfected individuals. Severe enthesopathy, dactylitis, and rapidly progressive joint destruction are seen, but axial involvement is very rare. This condition is prevented by or responds well to antiretroviral therapy.
LABORATORY AND RADIOGRAPHIC FINDINGS
There are no laboratory tests diagnostic of PsA. ESR and CRP are often elevated. A small percentage of patients may have low titers of rheumatoid factor or antinuclear antibodies. About 10% of patients have anti-CCP antibodies. Uric acid may be elevated in the presence of extensive psoriasis. HLA-B27 is found in 50–70% of patients with axial disease, but in ≤20% of patients with only peripheral joint involvement.
The peripheral and axial arthropathies in PsA show a number of radiographic features that distinguish them from RA and AS, respectively. Characteristics of peripheral PsA include DIP involvement, including the classic “pencil-in-cup” deformity; marginal erosions with adjacent bony proliferation (“whiskering”); small-joint ankylosis; osteolysis of phalangeal and metacarpal bone, with telescoping of digits; and periostitis and proliferative new bone at sites of enthesitis. Characteristics of axial PsA include asymmetric sacroiliitis. When compared with idiopathic AS, axial PsA manifests less zygapophyseal joint arthritis; nonmarginal, bulky, “comma”-shaped syndesmophytes that tend to be fewer and less symmetric and delicate than the marginal syndesmophytes of AS; fluffy hyperperiostosis on anterior vertebral bodies; severe cervical spine involvement, with a tendency to atlantoaxial subluxation but relative sparing of the thoracolumbar spine; and paravertebral ossification. Ultrasound and MRI both readily demonstrate enthesitis and tendon sheath effusions that can be difficult to assess on physical examination. A recent MRI study of 68 PsA patients found sacroiliitis in 35%, unrelated to B27 but correlated with restricted spinal movement.
DIAGNOSIS
Classification criteria for PsA were published in 2006 (Classification of Psoriatic Arthritis [CASPAR] criteria) that have been widely accepted (Table 384-2). The sensitivity and specificity of these criteria exceed 90%, and they are useful for early diagnosis. The criteria are based on the history, presence of psoriasis, characteristic peripheral or spinal joint symptoms, signs, and imaging. Diagnosis can be challenging when the arthritis precedes psoriasis, the psoriasis is undiagnosed or obscure, or the joint involvement closely resembles another form of arthritis. A high index of suspicion is needed in any patient with an undiagnosed inflammatory arthropathy. The history should include inquiry about psoriasis in the patient and family members. Patients should be asked to disrobe for the physical examination, and psoriasiform lesions should be sought in the scalp, ears, umbilicus, and gluteal folds in addition to more accessible sites; the finger and toe nails should also be carefully examined. Axial symptoms or signs, dactylitis, enthesitis, ankylosis, the pattern of joint involvement, and characteristic radiographic changes can be helpful clues. The differential diagnosis includes all other forms of arthritis, which can occur coincidentally in individuals with psoriasis. The differential diagnosis of isolated DIP involvement is short. Osteoarthritis (Heberden’s nodes) is usually not inflammatory; gout involving more than one DIP joint often involves other sites and may be accompanied by tophi; the very rare entity multicentric reticulohistiocytosis involves other joints and has characteristic small pearly periungual skin nodules; and the uncommon entity inflammatory osteoarthritis, like the others, lacks the nail changes of PsA. Radiography can be helpful in all of these cases and in distinguishing between psoriatic spondylitis and idiopathic AS. A history of trauma to an affected joint preceding the onset of arthritis is said to occur more frequently in PsA than in other types of arthritis, perhaps reflecting the Koebner phenomenon in which psoriatic skin lesions can arise at sites of the skin trauma.
THE CASPAR (CLASSIFICATION CRITERIA FOR PSORIATIC ARTHRITIS) CRITERIAa |
aSpecificity of 99% and sensitivity of 91%. bCurrent psoriasis is assigned 2 points; all other features are assigned 1 point. cPsoriatic skin or scalp disease present at the time of examination, as judged by a rheumatologist or dermatologist. dHistory of psoriasis in a first- or second-degree relative. eOnycholysis, pitting, or hyperkeratosis. fSwelling of an entire digit. gIll-defined ossification near joint margins, excluding osteophyte formation.
Source: From W Taylor et al: Arthritis Rheum, 54:2665, 2006.
UNDIFFERENTIATED AND JUVENILE-ONSET SPONDYLOARTHRITIS
Many patients, usually young adults, present with some features of one or more of the spondyloarthritides discussed above. Until recently, these patients were said to have undifferentiated spondylo- arthritis, or simply spondyloarthritis, as defined by the 1991 European Spondyloarthropathy Study Group criteria. For example, a patient may present with inflammatory synovitis of one knee, Achilles tendinitis, and dactylitis of one digit. Some of these patients may have ReA in which the triggering infection remains clinically silent. In some other cases, the patient subsequently develops IBD or psoriasis, or the process eventually meets criteria for AS. This diagnosis of undifferentiated SpA was also commonly applied to patients with inflammatory back pain, who did meet modified New York criteria for AS. Most of these would now be classified under the new category of axial SpA (Table 384-1).
Comparable to the classification criteria for axial symptoms, the ASAS has recently formulated criteria for peripheral SpA. This is intended to exclude patients with axial symptoms and thus to divide the universe of patients with SpA into axial and exclusively peripheral subsets. These criteria are shown in Table 384-3.
ASAS CRITERIA FOR PERIPHERAL SPONDYLOARTHRITISa |
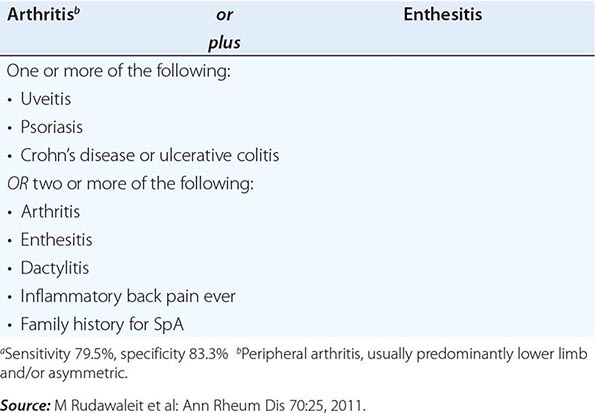
Approximately one-half of the patients with undifferentiated SpA are HLA-B27-positive, and thus the absence of B27 is not useful in establishing or excluding the diagnosis. In familial cases, which are much more frequently B27-positive, there is often eventual progression to classical AS.
In juvenile-onset SpA, which begins between ages 7 and 16, most commonly in boys (60–80%), an asymmetric, predominantly lower-extremity oligoarthritis and enthesitis without extraarticular features is the typical mode of presentation. The prevalence of B27 in this condition, which has been termed the seronegative enthesopathy and arthropathy (SEA) syndrome, is approximately 80%. Many, but not all, of these patients go on to develop AS in late adolescence or adulthood.
Management of undifferentiated SpA is similar to that of the other spondyloarthritides. Response to anti-TNF-α therapy has been documented, and this therapy is indicated in severe, persistent cases not responsive to other treatment.
Current pediatric textbooks and journals should be consulted for information on management of juvenile-onset SpA.
ENTEROPATHIC ARTHRITIS
HISTORIC BACKGROUND
A relationship between arthritis and IBD was observed in the 1930s. The relationship was further defined by the epidemiologic studies in the 1950s and 1960s and included in the concept of the spondylo- arthritides in the 1970s.
EPIDEMIOLOGY
Both of the common forms of IBD, ulcerative colitis (UC) and Crohn’s disease (CD) (Chap. 351), are associated with SpA. UC and CD both have an estimated prevalence of 0.05–0.1%, and the incidence of each is thought to have increased in recent decades. AS and peripheral arthritis are both associated with UC and CD. Wide variations have been reported in the estimated frequencies of these associations. In recent series, AS was diagnosed in 1–10%, and peripheral arthritis in 10–50% of patients with IBD. Inflammatory back pain and enthesopathy are common, and many patients have sacroiliitis on imaging studies.
The prevalence of UC or CD in patients with AS is thought to be 5–10%. However, investigation of unselected SpA patients by ileocolonoscopy has revealed that from one-third to two-thirds of patients with AS have subclinical intestinal inflammation that is evident either macroscopically or histologically. These lesions have also been found in patients with undifferentiated SpA or ReA (both enterically and urogenitally acquired).
Both UC and CD have a tendency to familial aggregation, more so for CD. HLA associations have been weak and inconsistent. HLA-B27 is found in up to 70% of patients with IBD and AS, but in ≤15% of patients with IBD and peripheral arthritis or IBD alone. Three alleles of the NOD2/CARD15 gene on chromosome 16 have been found in approximately one-half of patients with CD. These alleles are not associated with the spondyloarthritides per se. However, they are found significantly more often in (1) CD patients with sacroiliitis than in those without sacroiliitis, and (2) SpA patients with chronic inflammatory gut lesions than in those with normal gut histology. These associations are independent of HLA-B27. In addition to NOD2, over 100 other genes have been found to be associated with CD, UC, or both. Around 20 of these are also associated with AS.
PATHOLOGY
Available data for IBD-associated peripheral arthritis suggest a synovial histology similar to other spondyloarthritides. Association with arthropathy does not affect the gut histology of UC or CD (Chap. 351). The subclinical inflammatory lesions in the colon and distal ileum associated with SpA have been classified as either acute or chronic. The former resemble acute bacterial enteritis, with largely intact architecture and neutrophilic infiltration in the lamina propria. The latter resemble the lesions of CD, with distortion of villi and crypts, aphthoid ulceration, and mononuclear cell infiltration in the lamina propria.
PATHOGENESIS
Both IBD and SpA are immune-mediated, but the specific pathogenic mechanisms are poorly understood, and the connection between the two is obscure. The shared genetics evidently reflects shared pathogenic mechanisms. A number of rodent models showing various immune perturbations manifest both IBD and arthritis. Several lines of evidence indicate trafficking of leukocytes between the gut and the joint. Mucosal leukocytes from IBD patients have been shown to bind avidly to synovial vasculature through several different adhesion molecules. Macrophages expressing CD163 are prominent in the inflammatory lesions of both gut and synovium in the spondyloarthritides.
CLINICAL FEATURES
AS associated with IBD is clinically indistinguishable from idiopathic AS. It runs a course independent of the bowel disease, and in some patients, it precedes the onset of IBD, sometimes by many years. Peripheral arthritis may also begin before onset of overt bowel disease. The spectrum of peripheral arthritis includes acute self-limited attacks of oligoarthritis that often coincide with relapses of IBD, and more chronic and symmetric polyarticular arthritis that runs a course independent of IBD activity. The patterns of joint involvement are similar in UC and CD. In general, erosions and deformities are infrequent in IBD-associated peripheral arthritis, and joint surgery is infrequently required. Isolated destructive hip arthritis is a rare complication of CD, apparently distinct from osteonecrosis and septic arthritis. Dactylitis and enthesopathy are occasionally found. In addition to the ∼20% of IBD patients with SpA, a comparable percentage have arthralgias or fibromyalgia symptoms.
Other extraintestinal manifestations of IBD are seen in addition to arthropathy, including uveitis, pyoderma gangrenosum, erythema nodosum, and finger clubbing, all somewhat more commonly in CD than UC. The uveitis shares the features described above for PsA-associated uveitis.
LABORATORY AND RADIOGRAPHIC FINDINGS
Laboratory findings reflect the inflammatory and metabolic manifestations of IBD. Joint fluid is usually at least mildly inflammatory. Of patients with AS and IBD, 30–70% carry the HLA-B27 gene, compared with >85% of patients with AS alone and 50–70% of those with AS and psoriasis. Hence, definite or probable AS in a B27-negative individual in the absence of psoriasis should prompt a search for occult IBD. Radiographic changes in the axial skeleton are the same as in uncomplicated AS. Erosions are uncommon in peripheral arthritis but may occur, particularly in the metatarsophalangeal joints. Isolated destructive hip disease has been described.
DIAGNOSIS
Diarrhea and arthritis are both common conditions that can coexist for a variety of reasons. When etiopathogenically related, ReA and IBD-associated arthritis are the most common causes. Rare causes include celiac disease, blind loop syndromes, and Whipple’s disease. In most cases, diagnosis depends on investigation of the bowel disease.
SAPHO SYNDROME
The syndrome of synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) is characterized by a variety of skin and musculoskeletal manifestations. Dermatologic manifestations include palmoplantar pustulosis, acne conglobata, acne fulminans, and hidradenitis suppurativa. The main musculoskeletal findings are sternoclavicular and spinal hyperostosis, chronic recurrent foci of sterile osteomyelitis, and axial or peripheral arthritis. Cases with one or a few manifestations are probably the rule. The ESR is usually elevated, sometimes dramatically. In some cases, bacteria, most often Propionibacterium acnes, have been cultured from bone biopsy specimens and occasionally other sites. IBD was coexistent in 8% of patients in one large series. B27 is not associated. Either bone scan or computed tomography scan is helpful diagnostically. An MRI report described characteristic vertebral body corner cortical erosions in 12 of 12 patients. High-dose NSAIDs may provide relief from bone pain. A number of uncontrolled series and case reports describe successful therapy with pamidronate or other bisphosphonates. Response to anti-TNF-α therapy has also been observed, although in a few cases this has been associated with a flare of skin manifestations. Successful prolonged antibiotic therapy has also been reported. Recent reports suggest a possible autoinflammatory pathogenesis and successful treatment with the IL-1 receptor antagonist anakinra.
1Azathioprine, methotrexate, sulfasalazine, pamidronate, and thalidomide have not been approved for this purpose by the U.S. Food and Drug Administration at the time of publication.
385 | The Vasculitis Syndromes |
DEFINITION
Vasculitis is a clinicopathologic process characterized by inflammation of and damage to blood vessels. The vessel lumen is usually compromised, and this is associated with ischemia of the tissues supplied by the involved vessel. A broad and heterogeneous group of syndromes may result from this process, since any type, size, and location of blood vessel may be involved. Vasculitis and its consequences may be the primary or sole manifestation of a disease; alternatively, vasculitis may be a secondary component of another disease. Vasculitis may be confined to a single organ, such as the skin, or it may simultaneously involve several organ systems.
CLASSIFICATION
A major feature of the vasculitic syndromes as a group is the fact that there is a great deal of heterogeneity at the same time as there is considerable overlap among them. This heterogeneity and overlap in addition to a lack of understanding of the pathogenesis of these syndromes have been major impediments to the development of a coherent classification system for these diseases. Table 385-1 lists the major vasculitis syndromes. The distinguishing and overlapping features of these syndromes are discussed below.
VASCULITIS SYNDROMES |

PATHOPHYSIOLOGY AND PATHOGENESIS
Generally, most of the vasculitic syndromes are assumed to be mediated at least in part by immunopathogenic mechanisms that occur in response to certain antigenic stimuli. However, evidence supporting this hypothesis is for the most part indirect and may reflect epiphenomena as opposed to true causality. Furthermore, it is unknown why some individuals might develop vasculitis in response to certain antigenic stimuli, whereas others do not. It is likely that a number of factors are involved in the ultimate expression of a vasculitic syndrome. These include the genetic predisposition, environmental exposures, and the regulatory mechanisms associated with immune response to certain antigens. Although immune complex formation, antineutrophil cytoplasmic antibodies (ANCA), and pathogenic T lymphocyte responses (Table 385-2) have been among the prominent hypothesized mechanisms, it is likely that the pathogenesis of individual forms of vasculitis is complex and varied.
POTENTIAL MECHANISMS OF VESSEL DAMAGE IN VASCULITIS SYNDROMES |
Source: Adapted from MC Sneller, AS Fauci: Med Clin North Am 81:221, 1997.
PATHOGENIC IMMUNE-COMPLEX FORMATION
Deposition of immune complexes was the first and most widely accepted pathogenic mechanism of vasculitis. However, the causal role of immune complexes has not been clearly established in most of the vasculitic syndromes. Circulating immune complexes need not result in deposition of the complexes in blood vessels with ensuing vasculitis, and many patients with active vasculitis do not have demonstrable circulating or deposited immune complexes. The actual antigen contained in the immune complex has only rarely been identified in vasculitic syndromes. In this regard, hepatitis B antigen has been identified in both the circulating and deposited immune complexes in a subset of patients who have features of a systemic vasculitis, most notably in polyarteritis nodosa (see “Polyarteritis Nodosa”). Cryoglobulinemic vasculitis is strongly associated with hepatitis C virus infection; hepatitis C virions and hepatitis C virus antigen-antibody complexes have been identified in the cryoprecipitates of these patients (see “Cryoglobulinemic Vasculitis”).
The mechanisms of tissue damage in immune complex–mediated vasculitis resemble those described for serum sickness. In this model, antigen-antibody complexes are formed in antigen excess and are deposited in vessel walls whose permeability has been increased by vasoactive amines such as histamine, bradykinin, and leukotrienes released from platelets or from mast cells as a result of IgE-triggered mechanisms. The deposition of complexes results in activation of complement components, particularly C5a, which is strongly chemotactic for neutrophils. These cells then infiltrate the vessel wall, phagocytose the immune complexes, and release their intracytoplasmic enzymes, which damage the vessel wall. As the process becomes subacute or chronic, mononuclear cells infiltrate the vessel wall. The common denominator of the resulting syndrome is compromise of the vessel lumen with ischemic changes in the tissues supplied by the involved vessel. Several variables may explain why only certain types of immune complexes cause vasculitis and why only certain vessels are affected in individual patients. These include the ability of the reticuloendothelial system to clear circulating complexes from the blood, the size and physicochemical properties of immune complexes, the relative degree of turbulence of blood flow, the intravascular hydrostatic pressure in different vessels, and the preexisting integrity of the vessel endothelium.
ANTINEUTROPHIL CYTOPLASMIC ANTIBODIES (ANCA)
ANCA are antibodies directed against certain proteins in the cytoplasmic granules of neutrophils and monocytes. These autoantibodies are present in a high percentage of patients with active granulomatosis with polyangiitis (Wegener’s) and microscopic polyangiitis, and in a lower percentage of patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Because these diseases share the presence of ANCA and small-vessel vasculitis, some investigators have come to refer to them collectively as “ANCA-associated vasculitis.” However, as these diseases possess unique clinical phenotypes in which ANCA may be absent, it remains our opinion that granulomatosis with polyangiitis (Wegener’s), microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis (Churg-Strauss) should continue to be viewed as separate entities.
There are two major categories of ANCA based on different targets for the antibodies. The terminology of cytoplasmic ANCA (cANCA) refers to the diffuse, granular cytoplasmic staining pattern observed by immunofluorescence microscopy when serum antibodies bind to indicator neutrophils. Proteinase-3, a 29-kDa neutral serine proteinase present in neutrophil azurophilic granules, is the major cANCA antigen. More than 90% of patients with typical active granulomatosis with polyangiitis (Wegener’s) have detectable antibodies to proteinase-3 (see below). The terminology of perinuclear ANCA (pANCA) refers to the more localized perinuclear or nuclear staining pattern of the indicator neutrophils. The major target for pANCA is the enzyme myeloperoxidase; other targets that can produce a pANCA pattern of staining include elastase, cathepsin G, lactoferrin, lysozyme, and bactericidal/permeability-increasing protein. However, only antibodies to myeloperoxidase have been convincingly associated with vasculitis. Antimyeloperoxidase antibodies have been reported to occur in variable percentages of patients with microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis (Churg-Strauss), isolated necrotizing crescentic glomerulonephritis, and granulomatosis with polyangiitis (Wegener’s) (see below). A pANCA pattern of staining that is not due to antimyeloperoxidase antibodies has been associated with nonvasculitic entities such as rheumatic and nonrheumatic autoimmune diseases, inflammatory bowel disease, certain drugs, and infections such as endocarditis and bacterial airway infections in patients with cystic fibrosis.
It is unclear why patients with these vasculitis syndromes develop antibodies to myeloperoxidase or proteinase-3 or what role these antibodies play in disease pathogenesis. There are a number of in vitro observations that suggest possible mechanisms whereby these antibodies can contribute to the pathogenesis of the vasculitis syndromes. Proteinase-3 and myeloperoxidase reside in the azurophilic granules and lysosomes of resting neutrophils and monocytes, where they are apparently inaccessible to serum antibodies. However, when neutrophils or monocytes are primed by tumor necrosis factor α (TNF-α) or interleukin 1 (IL-1), proteinase-3 and myeloperoxidase translocate to the cell membrane, where they can interact with extracellular ANCA. The neutrophils then degranulate and produce reactive oxygen species that can cause tissue damage. Furthermore, ANCA-activated neutrophils can adhere to and kill endothelial cells in vitro. Activation of neutrophils and monocytes by ANCA also induces the release of proinflammatory cytokines such as IL-1 and IL-8. Adoptive transfer experiments in genetically engineered mice provide further evidence for a direct pathogenic role of ANCA in vivo. In contradiction, however, a number of clinical and laboratory observations argue against a primary pathogenic role for ANCA. Patients may have active granulomatosis with polyangiitis (Wegener’s) in the absence of ANCA; the absolute height of the antibody titers does not correlate well with disease activity; and patients with granulomatosis with polyangiitis (Wegener’s) in remission may continue to have high antiproteinase-3 (cANCA) titers for years (see below).
PATHOGENIC T LYMPHOCYTE RESPONSES AND GRANULOMA FORMATION
The histopathologic feature of granulomatous vasculitis has provided evidence to support a role of pathogenic T lymphocyte responses and cell-mediated immune injury. Vascular endothelial cells can express HLA class II molecules following activation by cytokines such as interferon (IFN) γ. This allows these cells to participate in immunologic reactions such as interaction with CD4+ T lymphocytes in a manner similar to antigen-presenting macrophages. Endothelial cells can secrete IL-1, which may activate T lymphocytes and initiate or propagate in situ immunologic processes within the blood vessel. In addition, IL-1 and TNF-α are potent inducers of endothelial-leukocyte adhesion molecule 1 (ELAM-1) and vascular cell adhesion molecule 1 (VCAM-1), which may enhance the adhesion of leukocytes to endothelial cells in the blood vessel wall.



