The Skin
Vijaya B. Reddy, M.D., M.B.A.
A variety of benign and malignant neoplasms as well as life-threatening inflammatory dermatoses can occur in children and require accurate diagnosis and timely management. Although any adult dermatologic disease can be seen in children, there are several conditions that are of clinical significance that occur with greater frequency, or at times exclusively, in children and neonates. This chapter, while covering the spectrum of dermatologic diseases, focuses specifically on the clinicopathologic features of the diseases encountered in children. A majority of the congenital diseases involving the skin are diagnosed clinically and rarely need biopsy; however, a substantial number of pediatric dermatologic diseases can only be diagnosed with specificity on histopathologic grounds. An algorithmic approach based on low-power pattern analysis is often the best place to start in the histopathologic evaluation of a skin biopsy (Table 26-1). As in other areas of pathology, clinicopathologic correlation is an integral part of the diagnostic process.
EMBRYOLOGY
The epithelial structures of the skin, namely, epidermis, folliculosebaceous units, and apocrine and eccrine sweat glands, are derived from the ectoderm. The dermis and its mesenchymal constituents, namely, vessels, smooth muscle, and nerve bundles, originate from the mesoderm.
There is a third component of skin that is composed of the migratory cells that originate at different sites and populate the skin. The melanocytes, Merkel cells, and Langerhans histiocytes form an integral part of the epidermis, while the mast cells and dendritic cells are present in the dermis. Melanocytes, Merkel cells, and perineural cells are neural crest derivatives. Mast cells and Langerhans cells are derived from mesenchymal precursors of bone marrow.
Skin development starts as a single layer of cells or periderm, which can be recognized in a 3-week-old embryo. There is progressive stratification of the epidermis, and by the end of the first trimester, several layers of glycogenrich cells can be seen in the epidermis. Cornification of the epidermis is completed during the 6th month of gestation (Figure 26-1). Defects in cornification (ichthyoses) can be diagnosed through fetal skin biopsies at this stage. At about 12 weeks, folliculosebaceous units and sweat glands begin as buds of basal cells that protrude into the mesenchyme of the dermis. Ectodermal dysplasias, which are characterized by absence of follicles and sweat glands, can be detected through fetal skin biopsies after the second trimester.
By the end of the first trimester, the dermoepidermal junction can be recognized, and at about 6 months, the dermal papillae become recognizable. The dermis, which begins as loosely arranged mesenchymal cells in a myxoid background, continues to be modified throughout the third trimester and beyond.
Because fetal skin biopsies have become increasingly useful in the diagnosis of genodermatoses, an understanding of the embryology of skin is critical not only in selecting the time of biopsy but also in interpretation of the biopsy findings.
NORMAL HISTOLOGY
Fetal skin is characterized by a virtually absent stratum corneum and an epidermis that is only a few cell layers thick. Depending on the gestational age, the dermis is relatively hypocellular and myxoid. Rudiments of adnexa including hair follicles and sweat glands can be identified starting from 20 to 24 weeks of gestation. In a premature baby, subcutaneous fat is virtually absent.
The dermis is thin in children compared with that in adults, with proportionately larger component of subcutaneous fat. With increasing age, the epidermis and the stratum corneum increase in thickness and the dermis becomes more compact and thick. Anatomic variations exist within the normal spectrum, and awareness of these features may prove helpful in localizing the site of biopsy when clinical information is lacking. These include numerous terminal hair follicles in the scalp, many vellus hair follicles and sebaceous lobules in facial skin, apocrine glands in the axilla and genitalia, and eccrine glands in acral skin. A prominent stratum granulosum and stratum corneum characteristic of chronic trauma are present in biopsies from the palm and sole.
TABLE 26-1 ALGORITHMIC APPROACH TO CUTANEOUS PATHOLOGY | |
|---|---|
|
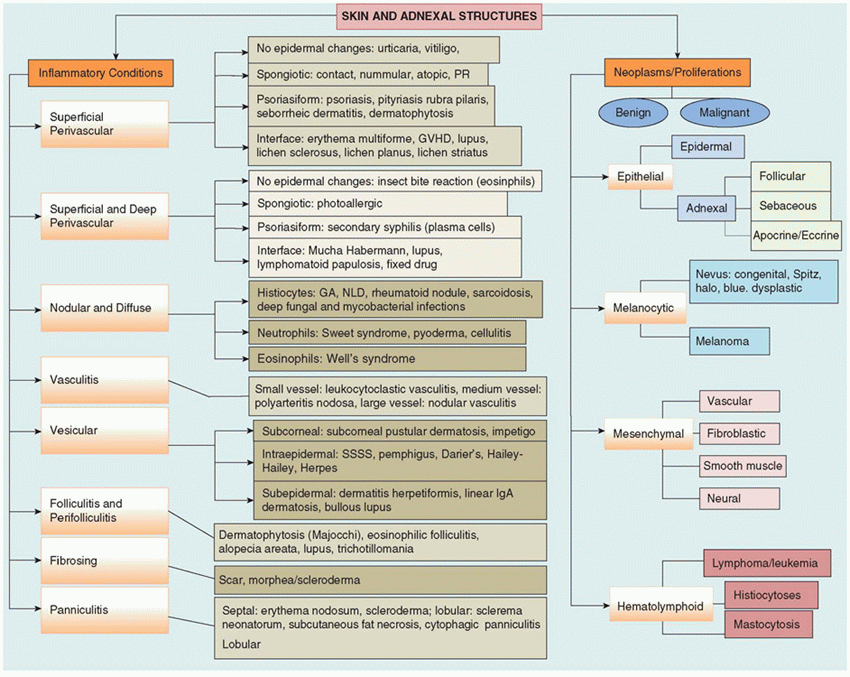
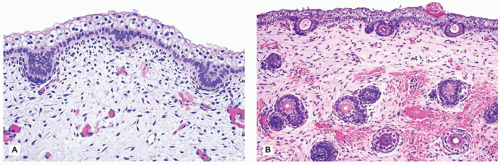 FIGURE 26-1 • A: Skin of an early, second trimester fetus showing stratification of epidermis, beginning of the follicular germs, and immature dermis. B: Skin from a 28-week fetus with stratum corneum and adnexal structures in a collagenized dermis. (Hematoxylin and eosin stain, original magnifications ×200.) |
BIOPSY TECHNIQUES
Skin samples can be obtained using various biopsy techniques, including punch, shave, excision, and curettage. Selection of the appropriate biopsy method depends on the clinical impression and the kind of information anticipated by the clinician.
Punch biopsy is generally the best choice for evaluation of inflammatory dermatoses. This technique allows the histologic examination of the full thickness of the skin including the subcutaneous fat. A 4-mm punch biopsy provides an adequate sample. In small children and cosmetically important areas, a 3-mm punch may be substituted. The area selected for biopsy should be a well-developed lesion and representative of the pathologic process.
Shave biopsy is the technique used in the evaluation of lesions that appear to be confined to the epidermis and superficial dermis and is best for the clinical diagnosis of keratoses and other benign neoplastic lesions. This technique is also used for suspected benign melanocytic lesions as well as for diagnostic confirmation of basal cell or squamous cell carcinoma prior to undertaking definitive surgical procedure.
Excisional biopsy is the technique of choice for suspected malignancies or atypical pigmented lesions. It generally allows for the evaluation of surgical margins, and as such, the lateral and deep margins should be inked before sectioning. Excisional biopsy or an incisional biopsy can also be used when panniculitis is clinically suspected.
Curettage is the technique some clinicians employ in examining clinically benign lesions. From a pathologist’s point of view, this is not a preferred method because the fragments of tissue so obtained are often small and superficial, precluding accurate analysis. Furthermore, if a clinically benign lesion turns out to be malignant on histologic examination, vital prognostic information such as invasion and thickness cannot be obtained since curetted fragments are difficult to orient.
Scrape preparation is useful in evaluation of viral vesicles, when cells are scraped off a vesicular or pustular lesion and analyzed after a quick stain.
Fine-needle aspiration biopsy is a popular method of biopsy in the evaluation of subcutaneous bumps and lumps. However, it requires an experienced cytopathologist for proper handling and interpretation of the material obtained.
SPECIMEN HANDLING
Routine Processing
Biopsy specimens should be placed immediately in a fixative. The fixative of choice for the majority of the specimens is 10% buffered formalin. Punch and shave biopsies larger than 3 mm in diameter should be bisected for optimal fixation as well as for appropriate plane of sectioning through the lesion, which is usually located in the center of the specimen. Excisional biopsies should be inked and sectioned at 2- to 3-mm intervals. Histologic sections cut at 3 to 5 µm are routinely stained with hematoxylin and eosin.
Special Processing
Specimens for direct immunofluorescence (IF) testing of bullous diseases are ideally obtained by biopsy of perilesional skin. A well-established lesion is best for suspected cases of lupus, whereas an early lesion is ideal for suspected case of vasculitis. Michel fixative is a good transport medium because the specimen can be safely tested for IF for up to 7 days. Alternatively, the specimen can be placed in normal saline and transported immediately to the laboratory. Frozen sections are incubated with fluorescein-labeled antibodies typically against IgG, IgA, IgM, C3, C1q, and fibrinogen and evaluated with IF microscope.
Electron microscopy may be of use in the diagnosis of undifferentiated neoplasms and can be invaluable in establishing the diagnosis of various types of epidermolysis bullosa and also in metabolic disorders like Fabry disease.
Specimens for electron microscopy should be fixed immediately in 2% glutaraldehyde or paraformaldehyde.
Specimens for electron microscopy should be fixed immediately in 2% glutaraldehyde or paraformaldehyde.
Skin and subcutaneous tissue may be used for cytogenetic analysis. Sterile specimens should be placed in a transport medium such as RPMI.
ALGORITHMIC APPROACH TO SPECIFIC DIAGNOSES
A systematic approach in analysis of biopsy sections allows for a smooth and accurate diagnosis. The pattern analysis method, which was introduced by Wallace H. Clark Jr., popularized by A. Bernard Ackerman and followed widely by most dermatopathologists today, involves the evaluation of sections at a scanning magnification. Examination at scanning magnification can be highly informative and usually helps in classifying a disease process as either proliferation/neoplastic or inflammatory. Table 26-1 is rather simplistic but offers the flavor for an algorithmic approach and includes representative dermatoses that occur in the pediatric patient. The following is a brief description of diseases grouped according to etiology and pathogenesis.
CONGENITAL DISEASES (GENODERMATOSES)
Genodermatoses are a large and diverse group of disorders presenting with cutaneous involvement and an underlying genetic defect. Only those genodermatoses that can be diagnosed in the fetus, neonate, or child will be discussed. It must be emphasized that some of these will be rarely seen by the pathologist, while others are not uncommon and are frequently included in the differential diagnosis.
Aplasia Cutis Congenita
Aplasia cutis congenita, or localized absence of skin, presents as a single or multiple skin defects typically involving the scalp. It presents at birth with a deep ulcer-like lesion, in which the subcutaneous fat is exposed. If the lesion occurs in utero, it manifests as a healed scar at birth. A sporadic lesion of aplasia cutis has no significant clinical consequences. More often, aplasia cutis may be a part of a variety of inheritable or noninheritable syndromes including trisomy 13, amniotic bands, and cardiac anomalies (1,2).
A histologic section of aplasia cutis shows a full-thickness skin defect with healing at the edges. A fully healed lesion shows scar with absence of adnexal structures. Formerly considered as synonymous, congenital absence of skin, characterized by absence of epidermis only, is now regarded as part of the epidermolysis bullosa group of disorders.
Ichthyosis
Ichthyoses are a heterogeneous group of disorders of epidermal cornification that are characterized by dryness and scaling of the skin. Ichthyoses are generally inherited, although acquired forms are described, especially in association with hematopoietic malignancies. The hereditary forms are divided into (a) the primary forms, which include ichthyosis vulgaris, recessive X-linked ichthyosis, epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma), classical lamellar ichthyosis, and nonbullous congenital ichthyosiform erythroderma; (b) ichthyosiform disorders such as Harlequin ichthyosis, erythrokeratoderma variabilis, and CHILD (congenital hemidysplasia with ichthyosiform erythrodermal and limb defects) syndrome; and (c) other related disorders of differentiation such as Darier disease, Hailey-Hailey disease, and porokeratosis (3).
Ichthyosis vulgaris is a common disorder, inherited in an autosomal dominant pattern that presents with fine white to larger scales involving large areas of the body but most prominent on the extensor surfaces of the extremities with relative sparing of the flexural areas. Histologically, there is moderate hyperkeratosis with a decreased or an absent granular layer and follicular plugging (Figure 26-2).
X-linked ichthyosis inherited as a recessive disorder presents early in infancy with large brown scales involving the entire body with accentuation on the neck and behind ears and relative sparing of the flexural areas. Histologically, there is hyperkeratosis with normal or thickened granular layer.
Bullous congenital ichthyosiform erythroderma or epidermolytic hyperkeratosis has an autosomal dominant pattern of inheritance and presents with generalized erythema and blistering at birth. Microscopic features include hyperkeratosis, a characteristic vacuolization of the cells in spinous and granular layers and prominent keratohyaline granules (Figure 26-3).
Lamellar ichthyosis is inherited as an autosomal recessive disorder and characterized by large platelike scales involving face, trunk, and extremities with a predilection for flexor areas. Microscopic changes are nonspecific and include hyperkeratosis with or without foci of parakeratosis and mild epidermal hyperplasia (Figure 26-4). Lamellar ichthyosis can present as a collodion baby, in which the infant is encased in a keratinous membrane and superficially resembles a harlequin fetus. However, the membrane is usually shed in 10 to 14 days, following which the clinical features of lamellar ichthyosis become apparent.
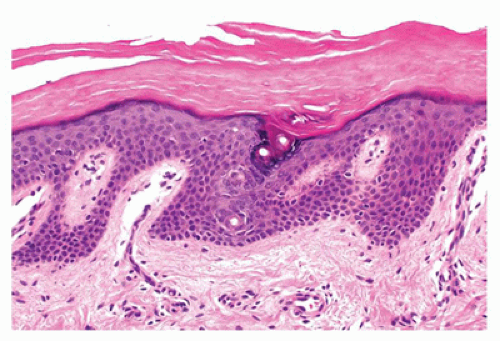 FIGURE 26-2 • Ichthyosis vulgaris showing hyperkeratosis and a prominent stratum corneum with a diminished granular cell layer. (Hematoxylin and eosin stain, original magnification ×200.) |
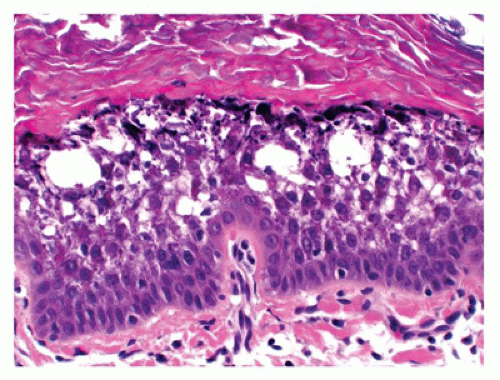 FIGURE 26-3 • Congenital bullous ichthyosiform erythroderma showing marked hyperkeratosis and epidermolytic hyperkeratosis with vacuolar degeneration of the stratum spinosum and granulosum, which is responsible for the formation of bullae. (Hematoxylin and eosin stain, original magnification ×200.) |
Nonbullous congenital ichthyosiform erythroderma is inherited as autosomal recessive disorder, is milder than lamellar ichthyosis, and has a more prominent erythrodermic component.
Fetal harlequin ichthyosis is an autosomal recessive disorder that can be fatal. Fetal skin biopsy can be diagnostic and shows massive hyperkeratosis. In utero, the massive hyperkeratosis interferes with normal development. At birth, the child is encased in a thick, fissured, scaly cast, associated with ectropion.
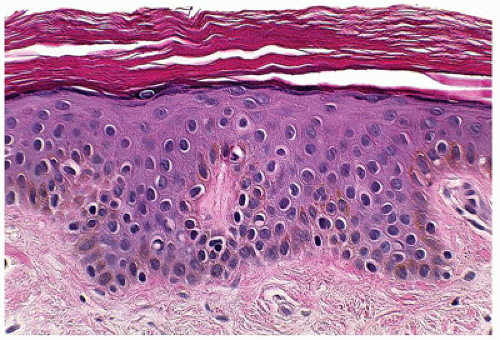 FIGURE 26-4 • Lamellar ichthyosis showing hyperkeratosis and mild psoriasiform changes. (Hematoxylin and eosin stain, original magnification ×200.) |
Darier Disease
Darier disease, also known as keratosis follicularis, is a relatively uncommon disease that is inherited in an autosomal dominant pattern. Darier disease gene has been localized to chromosome 12 (4). It typically presents in children aged 5 to 15 years as keratotic papules distributed in the seborrheic areas such as face, neck, and upper trunk (5). Oral mucosa and nails can also be involved (6,7). The histopathologic findings are characterized by suprabasal acantholysis covered by dyskeratotic cells (corps ronds) and parakeratosis (corps grains), in addition to papillomatous epidermal hyperplasia and hyperkeratosis (Figure 26-5). Occasionally, these lesions are centered on the hair follicles.
Most cases of Darier disease have a benign but protracted course with exacerbations during summer.
Hailey-Hailey Disease
Hailey-Hailey disease is an autosomal dominant genodermatosis that initially manifests typically only after puberty (late teens or early 20s). It is characterized by recurrent vesicles and erosions on the neck, axillae, and groin. Mucosal involvement is uncommon. Histologic features include suprabasal acantholysis resulting in a dilapidated brick wall-like appearance (Figure 26-6). Most cases of Hailey-Hailey disease have a fairly stable course. The cutaneous lesions are exacerbated by heat, humidity, and bacterial and candidal infections.
Porokeratosis
Porokeratosis is inherited as an autosomal dominant disorder that manifests in childhood and infancy as asymptomatic keratotic papules that enlarge progressively to form plaques with peripheral keratotic ridges. Four variants of porokeratosis can be seen in the pediatric population and include the classic plaque type of Mibelli, linear porokeratosis, porokeratosis palmaris, plantaris et disseminata, and punctate
porokeratosis that is limited to palms and soles. A fifth type, disseminated superficial actinic porokeratosis, is a disease of adulthood (8,9,10,11). Histopathologic features common to all types of porokeratoses include a cornoid lamella, which is a column of parakeratosis that corresponds to the peripheral keratotic ridges seen clinically (Figure 26-7). The cornoid lamella overlies an area of epidermal invagination where there is a diminished granular zone and vacuolated and dyskeratotic keratinocytes that correspond to an abnormal clone of keratinocytes. In porokeratosis of Mibelli, the epidermal invagination is more pronounced and deep compared with other types of porokeratosis.
porokeratosis that is limited to palms and soles. A fifth type, disseminated superficial actinic porokeratosis, is a disease of adulthood (8,9,10,11). Histopathologic features common to all types of porokeratoses include a cornoid lamella, which is a column of parakeratosis that corresponds to the peripheral keratotic ridges seen clinically (Figure 26-7). The cornoid lamella overlies an area of epidermal invagination where there is a diminished granular zone and vacuolated and dyskeratotic keratinocytes that correspond to an abnormal clone of keratinocytes. In porokeratosis of Mibelli, the epidermal invagination is more pronounced and deep compared with other types of porokeratosis.
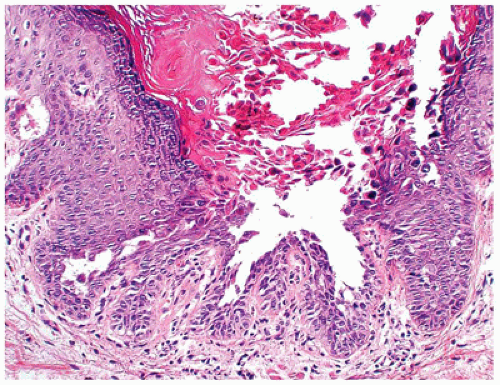 FIGURE 26-5 • Darier disease with focal intraepidermal acantholysis, dyskeratosis, and hyperkeratosis. (Hematoxylin and eosin stain, original magnification ×200.) |
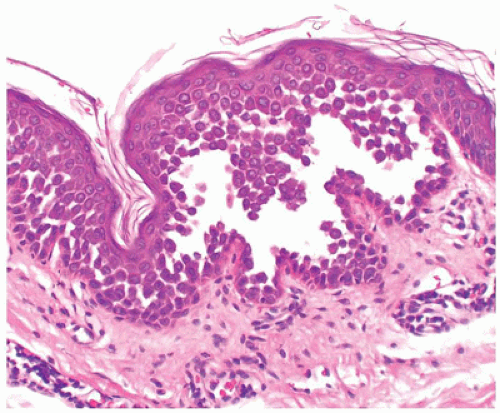 FIGURE 26-6 • Hailey-Hailey disease showing intraepidermal acantholysis with a dilapidated brick wall-like appearance. There is no significant dyskeratosis, which helps in the differential diagnosis from Darier disease. |
In addition to the inherited form, porokeratosis has been described in various immunosuppressive states including Crohn disease, renal transplantation, and HIV infection. Squamous cell carcinoma has been reported to develop in lesions of porokeratosis.
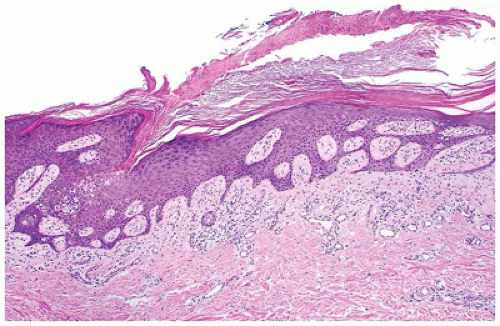 FIGURE 26-7 • Porokeratosis showing a column of parakeratosis that is inclined toward the center (cornoid lamella). Dyskeratotic keratinocytes may be seen at the base of the column. (Hematoxylin and eosin stain, original magnification ×100.) |
Restrictive Dermopathy
Restrictive dermopathy is an uncommon autosomal recessive disorder that presents with prematurity; rigid and tense skin with erosions, denudations, and multiple joint contractions; fixed facial expression; and perineal anomalies (Figure 26-8). Histologic features include a thickened epidermis with flattening of rete ridges and hyperkeratosis. The dermis is thin with absent elastic fibers, collagen bundles oriented parallel to the surface, and poorly developed adnexal structures (Figure 26-9). The disease is fatal, with most infants dying within weeks after birth. Abnormalities in collagen and abnormal synthesis of keratin have been proposed as the underlying defects.
Ectodermal Dysplasia
Ectodermal dysplasias form a large and heterogeneous group of genetic disorders that are characterized by developmental abnormalities in two or more major structures of ectodermal origin including hair, nails, teeth, sweat glands, and sebaceous glands. Ectodermal dysplasia syndromes are associated with abnormalities of nonectodermally derived structures with more than 100 syndromes encompassing all forms of Mendelian inheritance described clinically. The different disorders are differentiated based on the type of ectodermal defect (hair, teeth, nails, etc.), associated nonectodermal abnormalities, mode of inheritance, and underlying genetic defect. The two classic forms of ectodermal dysplasia include hidrotic and anhidrotic/hypohidrotic. The hidrotic form, with typically an autosomal dominant pattern of inheritance, is primarily a disorder of keratinization. It is characterized by hypotrichosis, dystrophic nails, and palmoplantar hyperkeratosis. The anhidrotic/hypohidrotic form is most commonly inherited as an X-linked recessive disorder localized to the Xq12-q13.1 region of X chromosome with full expression occurring only in men, who show the tetrad of anhidrosis or hypohidrosis, hypotrichosis, dental hypoplasia, characteristic facies, and frequently dystrophy of nails. In addition to aplasia and hypoplasia of sweat glands, the submucosal glands of the trachea and bronchus may be affected, leading to frequent respiratory infections. Histologically, both forms show hypoplasia of hair follicles and sebaceous glands. In addition, the anhidrotic form shows aplasia or hypoplasia of eccrine glands and occasionally of apocrine glands.
Focal Dermal Hypoplasia
Focal dermal hypoplasia was originally described by Libermann (12) as part of ectodermal and mesodermal dysplasia in association with osseous defects. The cutaneous aspects were first detailed by Goltz et al. (13). Because a majority of the patients are female, it has been assumed to have X-linked dominant mode of inheritance. Focal dermal hypoplasia is a multisystem condition in which developmental defects of skin are associated with ocular, dental, and skeletal system abnormalities. The clinical course is dictated by the extent of systemic involvement. The skin findings, which
are present from birth, consist of widely distributed asymmetric linear streaks of atrophy or hypoplasia of the skin, often with associated telangiectasia that follows Blaschko lines. Some lesions may present as soft yellow nodular outpouchings caused by herniation of fat through an atrophic dermis in linear array. Various mutations in X-linked PORCN, a putative regulator of Wnt signaling, have been identified in focal dermal hypoplasia (14,15).
are present from birth, consist of widely distributed asymmetric linear streaks of atrophy or hypoplasia of the skin, often with associated telangiectasia that follows Blaschko lines. Some lesions may present as soft yellow nodular outpouchings caused by herniation of fat through an atrophic dermis in linear array. Various mutations in X-linked PORCN, a putative regulator of Wnt signaling, have been identified in focal dermal hypoplasia (14,15).
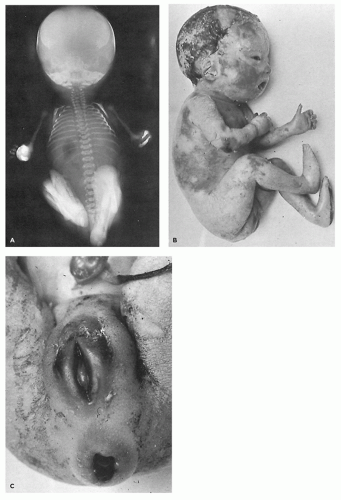 FIGURE 26-8 • A: Restrictive dermopathy showing severe contractures in the absence of overt bony abnormalities. B, C: The tight, shiny skin and characteristic fixation of the mouth and perineum. |
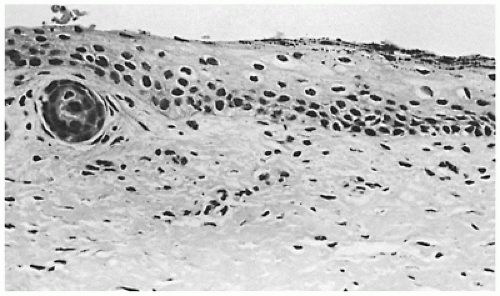 FIGURE 26-9 • Restrictive dermopathy without a stratum corneum, but the granular layer is prominent, indicating that the stratum corneum may have been lost in the postmortem interval. The rete ridges are flattened and most of the adnexal structures are atrophic. (Hematoxylin and eosin stain, original magnification ×250.) |
Histologic features of focal dermal hypoplasia include a marked decrease in the thickness of the dermis, with collagen distributed as thin fibrils rather than bundles, which may be interrupted by presence of adipocytes. The latter corresponds to the clinically apparent soft yellow nodules. The frequent presence of adipocytes high up in the dermis raises the possibility of nevus lipomatosus in the differential diagnosis. Nevus lipomatosus, however, lacks the collagen abnormalities and the frequent X-linked chromosomal abnormalities of focal dermal hypoplasia.
Epidermolysis Bullosa
Epidermolysis bullosa is a heterogeneous group of inherited disorders with variable modes of transmission, characterized by bullous lesions that develop spontaneously or secondary to minor trauma and include approximately 20 subtypes (16). Based on the presence or absence of scarring, mode of inheritance, cleavage plane of the blister, and the presence or absence of structural elements of skin, epidermolysis bullosa is traditionally divided into three major forms: simplex, junctional, and dystrophic (17,18).
Epidermolysis bullosa simplex, including Cockayne and Dowling-Meara forms, is typically transmitted in an autosomal dominant pattern, and is generally associated with good prognosis because the blisters heal without scar formation. Histologic sections show intraepidermal separation, generally within the basal cell layer. A periodic acid-Schiff (PAS) stain is helpful in localizing the level of cleavage above the basement membrane zone. Gene defects of keratins 5 and 14 are implicated and may be identified with IF mapping.
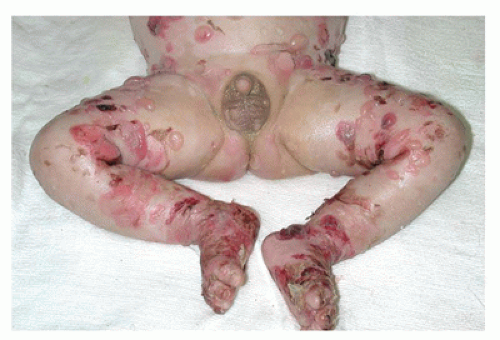 FIGURE 26-10 • Junctional epidermolysis bullosa in a 6-week-old infant showing extensive blistering and sloughing of the skin. (Courtesy of Sarah Stein, M.D., Department of Medicine, University of Chicago Medical Center.) |
Junctional epidermolysis bullosa is inherited as an autosomal recessive disorder and includes the fatal Herlitz type, in which blistering begins at birth and death occurs within the first 2 years (Figure 26-10), and the non-Herlitz type, which manifests similar to the Herlitz type but with a generally better overall prognosis. The cleavage plane occurs in the lamina lucida of the basement membrane at the dermoepidermal junction. Similar changes may involve the gastrointestinal, respiratory, and urinary tracts. Gene defects involving laminin 5 chain and collagen are identified.
Dystrophic epidermolysis bullosa includes the dominant form, which has a good prognosis, and the recessive form, which has a poor prognosis due to extensive erosions and ulcerations that heal with scarring. The level of cleavage is in the papillary dermis below the basement membrane (Figure 26-11). The principal gene defect involves collagen VII.
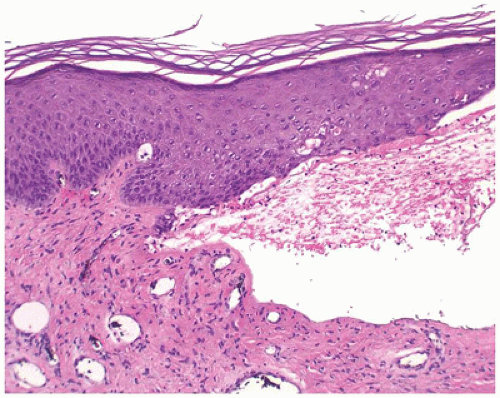 FIGURE 26-11 • Epidermolysis bullosa dystrophica has a subepidermal blister with mild dermal inflammation. (Hematoxylin and eosin stain, original magnification ×200.) |
Immunomapping studies are useful in localizing the cleavage plane and determining the presence, increase, or absence of the structural protein for which the gene is mutated in epidermolysis bullosa. These studies are essential for accurate classification of the type of epidermolysis bullosa, which in conjunction with clinical presentation forms the basis of prognostic information and genetic counseling. Furthermore, fetal skin biopsies during the third trimester can be diagnostic in the most severe forms of epidermolysis bullosa.
Incontinentia Pigmenti
Incontinentia pigmenti (Bloch-Sulzberger syndrome) is an X-linked dominant dermatosis that affects mostly women (19,20). Affected hemizygous male fetuses are generally thought to die in utero although recent literature suggests that some male individuals may show cutaneous and extracutaneous features of incontinentia pigmenti in a limited distribution that allow survival (21). The characteristic cutaneous manifestations evolve from crops of vesicles and bullae on the extremities arranged in linear or whorled pattern at birth or shortly thereafter that heal with hyperkeratotic verrucous lesions. As the verrucous lesions subside, characteristic streaks and whorls of hyperpigmentation develop, being most pronounced on the trunk. Faint hypochromic or atrophic lesions in a linear pattern may be seen on the lower extremities in some women and rarely in children (22).
Histologically, the vesicular stage is characterized by eosinophilic spongiosis and intraepidermal vesicle formation and eosinophil-rich dermal inflammatory cell infiltrate (Figure 26-12). The verrucous stage is characterized by hyperkeratosis and papillomatous epidermal hyperplasia with focal dyskeratosis. The hyperpigmented stage corresponds to numerous melanophages in the dermis as in any other postinflammatory pigmentary change.
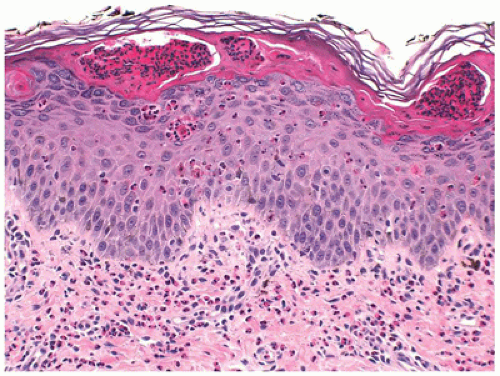 FIGURE 26-12 • Incontinentia pigmenti showing the initial skin changes with an intense eosinophilic infiltration within mildly spongiotic epidermis and the dermis. (Hematoxylin and eosin stain, original magnification ×200.) |
In approximately 80% of patients, systemic involvement, particularly of the central nervous system and the eye, and teeth abnormalities may be present. Although the skin manifestations are self-limiting, the clinical course is guided by the extent of systemic involvement.
Acrodermatitis Enteropathica
Acrodermatitis enteropathica is an autosomal recessive disorder characterized by defective intestinal absorption of zinc presenting with the triad of dermatitis, diarrhea, and alopecia in infancy at the time of weaning (23). Acquired acrodermatitis enteropathica-like syndromes can occur in exclusively breastfed preterm infants, infants who are fed on breast milk low in zinc, infants with organic acid urea, and any other acquired zinc deficiency states including human immunodeficiency virus infection. Cutaneous manifestations are characterized by vesiculobullous lesions with acral and periorificial distribution. The histopathologic findings include intraepidermal bulla with epidermal necrosis or spongiosis and superficial perivascular mixed inflammatory cell infiltrate. A wellestablished lesion shows parakeratosis, marked pallor and ballooning of keratinocytes, and a markedly diminished granular zone. The histologic changes can be identical to those seen in glucagonoma syndrome and pellagra, conditions associated with nutritional deficiencies of factors essential for normal maturation and metabolism of epidermal keratinocytes.
NONINFECTIOUS ACQUIRED VESICULOBULLOUS DISEASES
Linear IgA Bullous Dermatosis
Linear IgA bullous dermatosis, also known as chronic bullous dermatosis of childhood, presents with large tense bullae in prepubertal children often younger than 5 years of age. The lesions are widespread in distribution and vesicles and bullae, sometimes arranged like a string of pearls, occur at the periphery of a healing lesion. Areas of predilection include the lower part of the trunk, including the groin and genitalia and perioral areas. Rare cases have been described in neonates. Microscopic features are essentially indistinguishable from dermatitis herpetiformis and consist of neutrophilic microabscesses at the tips of dermal papillae in early lesions and subepidermal bulla filled with neutrophils or eosinophils in well-established lesions (Figure 26-13). Direct IF testing shows a distinct linear pattern of staining at the basement membrane zone with IgA, in sharp contrast to the granular IgA deposits seen in dermatitis herpetiformis. Direct IF testing is crucial in differentiating lupus erythematosus and chronic bullous dermatosis of childhood from other childhood bullous diseases like bullous pemphigoid and lichen planus. Chronic bullous disease of childhood generally has a benign course with spontaneous remission before puberty. Rare cases heal with scarring when the
disease process seems to overlap with childhood cicatricial pemphigoid, which some consider to be another morphologic expression of linear IgA dermatosis of childhood and adults. Linear IgA dermatosis of both children and adults are also similar, both IgA1-mediated diseases (24) with some cases of chronic bullous dermatosis of childhood relapsing into adulthood. Distinction of linear IgA bullous disease from dermatitis herpetiformis is important because the former is not typically associated with gluten-sensitive enteropathy.
disease process seems to overlap with childhood cicatricial pemphigoid, which some consider to be another morphologic expression of linear IgA dermatosis of childhood and adults. Linear IgA dermatosis of both children and adults are also similar, both IgA1-mediated diseases (24) with some cases of chronic bullous dermatosis of childhood relapsing into adulthood. Distinction of linear IgA bullous disease from dermatitis herpetiformis is important because the former is not typically associated with gluten-sensitive enteropathy.
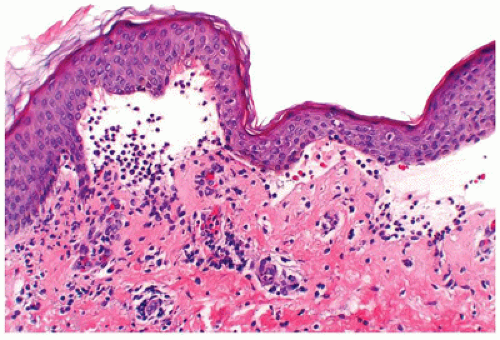 FIGURE 26-13 • Linear IgA dermatosis with a subepidermal blister with neutrophils. |
Dermatitis Herpetiformis
Dermatitis herpetiformis presents as an intensely pruritic papulovesicular eruption that is typically distributed on the scalp, the extensor aspects of extremities, and the back. The lesions may be grouped in herpetiform fashion and symmetrical in distribution. They are characterized by small papules and tense vesicles that rupture easily (25). Although dermatitis herpetiformis generally manifests as a skin disease, approximately 75% to 90% of the children with this disorder have an associated gluten-sensitive enteropathy and a high frequency of HLA antigens, including HLA B8, DR3, and DqW2 (26,27). Histologic sections of a papular lesion show the characteristic neutrophilic microabscesses at the tips of the dermal papillae (Figure 26-14). Sections of a clinically apparent vesicle show a subepidermal bulla filled with neutrophils and a varying mixture of eosinophils and fibrin. Microabscesses are present at the edge of the blister. Direct IF testing is positive for granular deposits of IgA at the tips of dermal papillae in almost all patients (28). A gluten-free diet is effective in controlling the intestinal and cutaneous manifestations in most children.
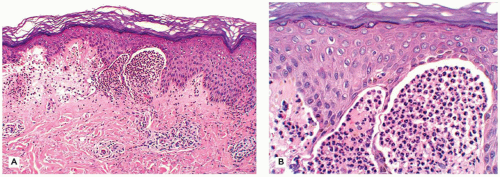 FIGURE 26-14 • Dermatitis herpetiformis. A, B: Subepidermal separation with neutrophilic microabscesses at the tips of dermal papillae. Differentiation from linear IgA dermatosis is possible only on IF studies. (Hematoxylin and eosin stains, original magnification ×200 (A), ×400 (B).) |
Herpes Gestationis
Pemphigoid gestationis (herpes gestationis) is a rare acquired autoimmune bullous disease that affects pregnant women most commonly during the second trimester (29) and, in a small percentage of cases, can be transmitted to the neonates born to these women. The affected neonate may present with macules or papulovesicular or bullous lesions at birth or shortly thereafter. In neonates, the condition is transient, with complete resolution of the lesions occurring within a month, and it is attributed to the transplacental transfer of maternal antibodies (30). Studies have failed to show significant association between pemphigoid gestationis and increased incidence and risk of fetal morbidity or mortality. Histopathologic and IF findings may be identical to those seen in bullous pemphigoid and consist of subepidermal bulla with eosinophils and linear deposits of C3 and IgG at the basement membrane zone. Additionally, sera from patients with pemphigoid gestationis like those with bullous pemphigoid are positive for antibodies against a 180-kD epidermal antigen (31).
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is an acquired form of epidermolysis bullosa that is seen more commonly in adults but can sometimes be seen in children (32,33). Clinically, it may resemble the autosomal dominant dystrophic type of epidermolysis bullosa manifesting with tense blisters on the extensor aspects that heal with scarring and milia formation. However, epidermolysis bullosa acquisita is an autoimmune disease characterized by IgG antibodies to type VII collagen of the basement membrane (34,35).
Light microscopic and IF findings are indistinguishable from bullous pemphigoid, which can also be seen in children (36). Both conditions are characterized by subepidermal bulla with linear deposits of IgG and C3 at the basement membrane zone. Indirect IF studies localize the IgG deposits to the roof of salt-split skin, in which the cleavage runs through the lamina lucida of the basement membrane in pemphigoid and the floor (beneath the lamina lucida) in epidermolysis bullosa acquisita.
Erythema Multiforme, Stevens-Johnson Syndrome, and Toxic Epidermal Necrolysis
Erythema multiforme, Stevens-Johnson (S-J) syndrome, and toxic epidermal necrolysis (TEN) (Lyell syndrome) form the clinical and histopathologic spectrum of a potentially lifethreatening group of disorders characterized by epidermal necrosis with formation of bullae, which can involve a large part of the skin surface and mucosa. The high mortality rate in patients with TEN is directly related to the resultant fluid loss and sepsis. S-J syndrome is more common in childhood than erythema multiforme or TEN.
Erythema multiforme is distinguished clinically by the characteristic iris or targetoid lesions that can occur on any part of the body but most commonly on the palms and soles. Erythematous and purpuric macules that progress to flaccid bullae and detach from the underlying dermis are characteristic of S-J syndrome and TEN. The detachment is extensive in TEN while mucosal involvement is more prominent in S-J syndrome.
The majority of cases of erythema multiforme in children are etiologically related to herpes simplex virus (HSV) infection; other viral infections including Epstein-Barr virus and orf and mycoplasma infections have also been implicated (37). Drugs such as sulfonamides and penicillins play an important role, especially in the more severe S-J syndrome and TEN (38,39). No cause can be identified in a significant number of cases.
Histopathologic features include interface dermatitis with vacuolar alteration of the basal cell layer and mild perivascular infiltrate of lymphocytes, which are also present along the dermoepidermal junction. The histologic hallmark of this group of diseases is the necrotic keratinocyte, which may be few in milder forms and numerous with confluent areas of necrosis in more established lesions (Figure 26-15). In TEN, full-thickness epidermal necrosis leads to subepidermal separation and loss of epidermal surface with the eroded clinical appearance of skin originally described by Lyell (Figure 26-16). An unaltered stratum corneum in skin biopsies attests to the acute nature of the assault on the skin. Immune complex-mediated reactions of types III and IV and helper T-cell-mediated immunoreactions are believed to play a role in the pathogenesis of erythema multiforme/TEN (40). Erythema multiforme/S-J syndrome/TEN are potentially life-threatening disorders that require hospitalization, withdrawal of recent drugs, and supportive care. Potential infectious causes should be sought and treated. The benefits of specific treatment
including corticosteroids and intravenous administration of immunoglobulin are still under debate and further investigation (41,42,43,44).
including corticosteroids and intravenous administration of immunoglobulin are still under debate and further investigation (41,42,43,44).
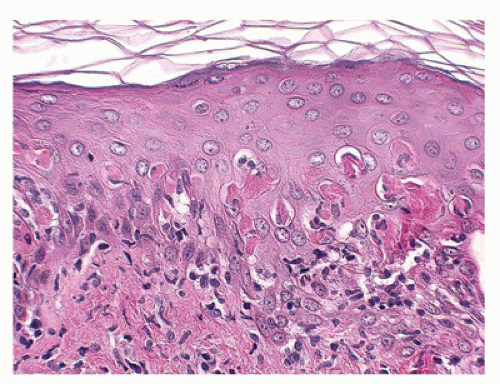 FIGURE 26-15 • Erythema multiforme with basket-weave orthokeratosis, vacuolar alteration of the basal cell layer, necrotic keratinocytes, and mild superficial perivascular inflammation. (Hematoxylin and eosin stain, original magnification ×400.) |
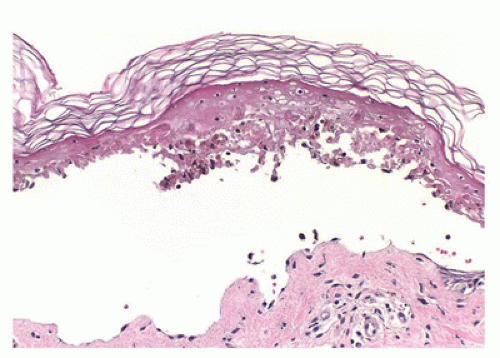 FIGURE 26-16 • TEN with full-thickness epidermal necrosis with separation at the dermoepidermal junction and sparse inflammatory cell infiltrate. An unaltered cornified layer attests to the acuteness of the event. (Hematoxylin and eosin stain, original magnification ×400.) |
MISCELLANEOUS NONINFECTIOUS VESICULOPUSTULAR DISEASES
A variety of benign vesiculopustular diseases are commonly seen in neonates and infants. It is important to differentiate these conditions from the more serious vesiculopustular diseases that can affect children (45).
Erythema Toxicum Neonatorum
Erythema toxicum neonatorum (toxic erythema of newborn) is an asymptomatic, transient, self-limiting, common eruption that occurs in the first 24 to 48 hours of life of fullterm newborns. The lesions are characterized by macules, papules, and tiny pustules that can affect any part of the body but favor the trunk and proximal extremities. Classical clinical presentation rarely requires a skin biopsy that would reveal eosinophils in the pilosebaceous units and differentiate erythema toxicum neonatorum from other neonatal pustular dermatoses including incontinentia pigmenti (46,47).
TRANSIENT NEONATAL PUSTULAR MELANOSIS
Transient neonatal pustular melanosis is a benign, self-limiting condition that predominantly affects black infants. The skin eruption begins as superficial sterile pustules that rupture easily and typically heal with hyperpigmented macules with collarettes of fine scale. Similarities between transient neonatal pustular melanosis and erythema toxicum neonatorum are emphasized by some authors who proposed “sterile transient neonatal pustulosis” as a unifying term (48). However, the pustules in transient neonatal pustular melanosis show abundance of neutrophils.
Acropustulosis of Infancy
Acropustulosis of infancy presents as recurrent crops of pruritic vesicles and pustules on distal extremities with predilection for palms and soles, primarily in black infants during the first year of life. Most cases show spontaneous resolution by the age of 2 years (49). Smears from the pustule and histologic sections of the subcorneal pustules show abundant neutrophils.
ECZEMATOUS DERMATITIS
“Eczema” is the term often used to describe erythematous, scaling vesicular lesions with serum crust. Eczematous dermatitis is characterized histologically by epidermal spongiosis and, therefore, is often referred to interchangeably as spongiotic dermatitis. A specific diagnosis is based on clinical history, morphologic appearance, and distribution of lesions. This group of disorders includes nummular dermatitis, contact dermatitis, dyshidrotic dermatitis, and atopic dermatitis.
Nummular Dermatitis
Nummular dermatitis is characterized by coin-shaped, pruritic, erythematous, scaly crusted plaques on the extensor aspect of the extremities. It is believed to be a manifestation of xerosis and is more commonly seen in older patients.
Atopic Dermatitis
Atopic dermatitis is an inherited chronic pruritic skin disease and is the most common skin disease seen in children, with an estimated incidence of as high as 20% (50). About one-third of the cases are diagnosed before the age of 1 year and before 5 years of age in vast majority of patients (51). Sites of predilection are the face in young infants, extensor surfaces of extremities in children younger than 1 year of age, and the popliteal and antecubital fossae, face, and neck in older children and adolescents. The major abnormality in this disease appears to be the overproduction of allergenspecific IgE, and some authors suggest that demonstration of such antibodies be a requisite for the diagnosis of atopic dermatitis (52). Cytokines, T cells, and antigen-presenting cells in addition to abnormalities of skin barrier appear to play a role in the pathogenesis (53).
Contact Dermatitis
Contact dermatitis includes primary irritant dermatitis and allergic contact dermatitis. Primary irritant dermatitis is frequently seen in children on the cheeks caused by saliva, extremities in response to harsh soaps or detergents, and the diaper area from toiletries (54). Allergic contact dermatitis presents with pruritic, edematous papules, plaques, and occasionally vesicles 12 to 24 hours after exposure to an allergen such as poison ivy, fragrances, nickel, and rubber compounds (55). Allergic contact dermatitis occurs more frequently in children with atopic tendencies (56).
Dyshidrotic Dermatitis
Dyshidrotic dermatitis (pompholyx) typically presents with numerous, pinpoint, recurrent, pruritic vesicles along the sides of the fingers and toes and on palms and soles that usually last a few weeks and frequently relapse.
Histopathology of Spongiotic Dermatitis
Irrespective of the specific type of disease, spongiotic dermatitis shows a similar spectrum of changes. In the acute phase, there is epidermal spongiosis, sometimes marked, with vesiculation (Figure 26-17). In the subacute phase, the spongiosis is milder, but associated parakeratosis with plasma cells, neutrophils, eosinophils, and epidermal hyperplasia may be present. In the chronic phase, the spongiosis is mild to absent, but changes of chronicity are reflected in a
hyperkeratotic cornified layer, marked epidermal hyperplasia, and fibrotic papillary dermis. Superficial perivascular lymphohistiocytic infiltrate is present to varying degrees in all the phases of spongiotic dermatitis.
hyperkeratotic cornified layer, marked epidermal hyperplasia, and fibrotic papillary dermis. Superficial perivascular lymphohistiocytic infiltrate is present to varying degrees in all the phases of spongiotic dermatitis.
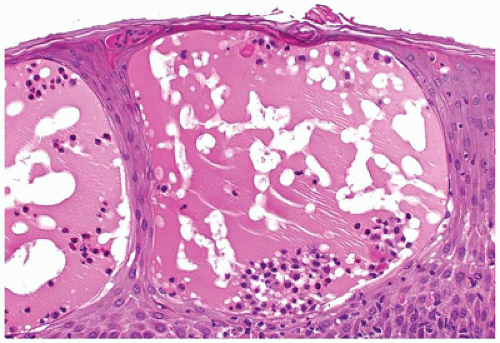 FIGURE 26-17 • Eczematous dermatitis showing marked epidermal spongiosis with formation of intraepidermal vesicles and moderate perivascular mixed inflammation. (Hematoxylin and eosin stain, original magnification ×400.) |
NONINFECTIOUS PAPULOSQUAMOUS DERMATOSES
This includes a group of diverse disorders characterized by papular and scaling lesions and associated epidermal proliferation. Approximately 10% of the patients seen in a pediatric dermatology clinic present with papulosquamous skin disorders (57). The following is a brief discussion of the more common dermatoses traditionally regarded as the papulosquamous dermatoses.
Psoriasis Vulgaris
Psoriasis vulgaris accounts for 4% of all dermatoses encountered in children younger than the age of 16 years, (58) and in about 30% of the patients, psoriasis manifests in the first or the second decade of life (59). Of the various forms of psoriasis, namely, plaque type, guttate, pustular and erythrodermic psoriasis, plaque type is the most common one seen in children followed by guttate psoriasis (60). Pustular psoriasis and psoriatic arthropathy are less common in children (61). The clinical presentation is characterized by asymptomatic scaly erythematous plaques in the plaque type and by slightly pruritic small red drop-like scaly lesions in guttate psoriasis. Silvery scales that, on scraping, leave pinpoint areas of bleeding (Auspitz sign) are typical of psoriasis. Lesions are distributed in a bilaterally symmetrical pattern with predilection for scalp and extensor aspects of extremities. Involvement of face is more common in children than in adults and needs to be distinguished from atopic dermatitis. Similarly, psoriasis may involve the diaper area in up to 13% of patients where it must be differentiated from infantile seborrheic dermatitis and other causes of diaper dermatitis (62). Classic histologic features of psoriasis include confluent parakeratosis with neutrophils (Munro microabscesses), regular elongation of epidermal rete with thin suprapapillary plates, dilated vessels in dermal papillae, and mild superficial perivascular inflammation (Figure 26-18). Dermatophytes can produce psoriasiform dermatitis.
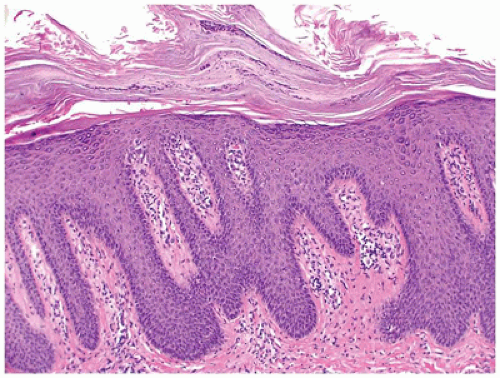 FIGURE 26-18 • Psoriasis is characterized by confluent parakeratosis and regular epidermal hyperplasia. (Hematoxylin and eosin stain, original magnification ×200.) |
Psoriasis, a multifactorial disorder with a genetic basis, typically runs a chronic course with remissions and flare-ups.
Seborrheic Dermatitis
A chronic dermatosis of unknown cause, seborrheic dermatitis is quite common in infants aged 2 to 10 weeks and in adolescents. In infants, seborrheic dermatitis begins as an erythematous scaly rash typically involving the scalp, face, and diaper area. In adolescents, it appears as a dry fine exfoliation of the scalp (dandruff) and expands to the face with the clinical features sometimes overlapping with those of psoriasis.
Histopathologic features overlap with psoriasis and spongiotic dermatitis and consist of epidermal hyperplasia and spongiosis with exocytosis and patchy parakeratosis, which is often present at the openings of the follicular infundibula. A mild superficial perivascular lymphohistiocytic inflammation is present in the dermis.
Infantile seborrheic dermatitis may clinically mimic Langerhans cell histiocytosis (LCH), which is a potentially serious disorder.
Lichen Planus
More commonly a disease of adulthood, lichen planus, generally a self-limiting pruritic eruption, is generally considered uncommon in children (63). However, children of South Asian subcontinent appear to be more susceptible to developing lichen planus (64). The clinical appearance of the eruption is distinctive and consists of flat-topped violaceous papules involving flexor aspects of the extremities and lower back. Lichen planus can also involve hair, nails, and mucous membranes in a significant number of cases. The histologic features are distinctive and consist of hyperkeratosis,
hypergranulosis, irregular epidermal hyperplasia, and a band-like lymphohistiocytic infiltrate that obscures the dermoepidermal junction (lichenoid dermatitis) where there are vacuolar alterations and colloid bodies (Figure 26-19). Melanophages are seen in the infiltrate in older lesions.
hypergranulosis, irregular epidermal hyperplasia, and a band-like lymphohistiocytic infiltrate that obscures the dermoepidermal junction (lichenoid dermatitis) where there are vacuolar alterations and colloid bodies (Figure 26-19). Melanophages are seen in the infiltrate in older lesions.
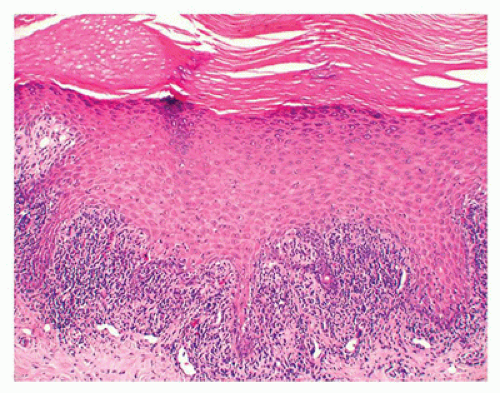 FIGURE 26-19 • Lichen planus showing hyperkeratosis, hypergranulosis, irregular epidermal hyperplasia, and a band-like lymphohistiocytic infiltrate that obscures the dermoepidermal junction. (Hematoxylin and eosin stain, original magnification ×400.) |
The etiology of lichen planus is unknown in most cases, whereas in others, various drugs have been implicated.
Pityriasis Rosea
Pityriasis rosea is an acute, self-limiting papulosquamous eruption appearing in children, especially adolescents, up to 45% of the time (65). It typically presents with a single large scaly plaque, the herald patch on the trunk that is followed within a week by more disseminated smaller oval scaly pink papules along the lines of skin cleavage. In addition to the trunk, the neck and proximal extremities may be involved. Histologic sections show focal parakeratosis, focal spongiosis, and a mild superficial perivascular lymphohistiocytic infiltrate. Extravasated red blood cells are often present in the papillary dermis and may extend into the epidermis (Figure 26-20). Biopsy of the herald patch also shows epidermal hyperplasia and denser infiltrate of inflammatory cells. A viral etiology has been suspected for a long time, and in recent years, viruses such as human herpes virus 7 and parvovirus have been implicated in the etiology (66). Most cases of pityriasis rosea resolve within 6 to 12 weeks with no specific treatment.
Pityriasis Rubra Pilaris
Pityriasis rubra pilaris is a chronic follicular-based erythematous papular eruption of unknown etiology that can manifest in children (67). Although most cases are acquired, a familial form is also recognized. Keratoderma of the palms and soles develops in a majority of the affected children and about 40% show cephalic involvement. Histologic findings include epidermal hyperplasia, alternating hyperkeratosis and parakeratosis oriented in both vertical and horizontal directions, and a mild superficial perivascular lymphocytic infiltrate. Follicular plugging is present in biopsies of the follicular papules (Figure 26-21).
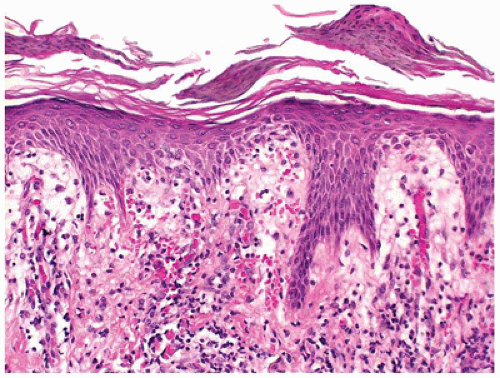 FIGURE 26-20 • Pityriasis rosea showing patchy parakeratosis (mounding parakeratosis) and focal spongiosis and extravasated red cells and inflammatory cells in the superficial dermis. (Hematoxylin and eosin stain, original magnification ×200.) |
Spontaneous resolution of the cutaneous rash is expected in a majority of the cases within 2 to 3 years, whereas recurrences and a protracted course may occur in others.
Pityriasis Lichenoides
Pityriasis lichenoides is a self-limiting cutaneous eruption of unknown cause that can occur in pediatric patients, commonly during the first decade of life (68). The cutaneous eruption may be delineated along a spectrum including an acute more severe form pityriasis lichenoides et varioliformis acuta (PLEVA, Mucha-Habermann disease) and a chronic milder form pityriasis lichenoides chronica. Transitional forms in between the two extremes are
recognized in children. In addition, a more severe but rare variant, the acute febrile ulceronecrotic form, which is more common in children, has also been described (69). PLEVA is characterized by an extensive papular, papulonecrotic, and, occasionally, vesiculopustular eruption on the trunk and proximal extremities that resolves within a few weeks. As the older lesions resolve, crops of newer lesions continue to appear, and the overall course may be protracted to several months. The ulceronecrotic form is characterized by large coalescing ulceronecrotic nodules and plaques associated with high fever.
recognized in children. In addition, a more severe but rare variant, the acute febrile ulceronecrotic form, which is more common in children, has also been described (69). PLEVA is characterized by an extensive papular, papulonecrotic, and, occasionally, vesiculopustular eruption on the trunk and proximal extremities that resolves within a few weeks. As the older lesions resolve, crops of newer lesions continue to appear, and the overall course may be protracted to several months. The ulceronecrotic form is characterized by large coalescing ulceronecrotic nodules and plaques associated with high fever.
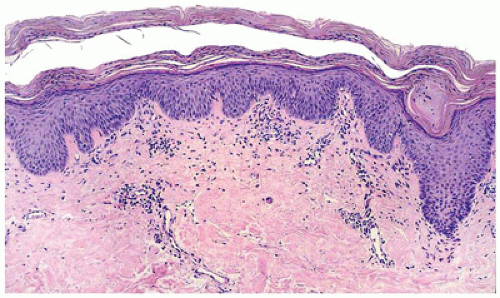 FIGURE 26-21 • Pityriasis rubra pilaris is characterized by mild epidermal hyperplasia with alternating layers of hyperkeratosis and parakeratosis and follicular plugging. (Hematoxylin and eosin stain, original magnification ×200.) |
The chronic form of pityriasis lichenoides chronica is characterized by recurrent crops of reddish-brown papules with an adherent scale that typically resolve within 3 to 6 weeks without scarring. Transient postinflammatory pigmentary changes may occur.
Histopathologic findings in pityriasis lichenoides include interface dermatitis with parakeratosis, epidermal spongiosis, necrotic keratinocytes, and a perivascular lymphocytic infiltrate. Papillary dermal edema and extravasated red cells may be present. In PLEVA, the inflammatory cell infiltrate is dense and deep, and spongiosis and epidermal necrosis are more marked with eventual erosion or ulceration of the epidermis with overlying parakeratotic scale crust containing neutrophils (Figure 26-22).
Clinical and histopathologic findings may show some overlap with lymphomatoid papulosis, a benign, recurrent self-healing dermatosis that falls within the spectrum of CD30-positive cutaneous lymphoproliferative disorders (70). Studies have shown T-cell clonality in pityriasis lichenoides, especially in the acute form (71,72), suggesting that host immune reaction prevents further progression to lymphoma. Although evolution to cutaneous T-cell lymphoma has been reported, in a long-term follow-up study of 89 children with pityriasis lichenoides, the clinical course was essentially benign, with no evolution into lymphomatoid papulosis or lymphoma (73).
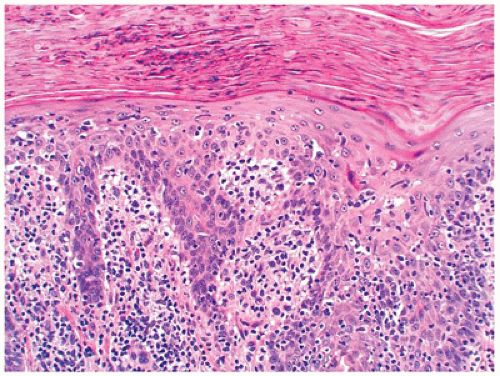 FIGURE 26-22 • PLEVA with mounds of parakeratosis containing neutrophils and interface dermatitis with vacuolar alteration of the basal cell layer and necrotic keratinocytes. (Hematoxylin and eosin stain, original magnification ×200.) |
Papular Acrodermatitis of Childhood
Papular acrodermatitis of childhood, or Gianotti-Crosti syndrome, is a self-limiting papular and papulovesicular eruption involving the face, extremities, and buttocks of children aged 2 to 6 years with an underlying viral infection. Since the original description of the cases in association with hepatitis B infection, a variety of other viruses including Coxsackie, Epstein-Barr, parainfluenza, pox, parvovirus B19, and HIV and certain bacteria as well as immunizations have been shown to be associated with similar cutaneous eruptions. In recent years, there has been a striking shift from HBV to EBV as the most common cause, although the exact mechanism of how the infectious agents cause the cutaneous eruption continues to reside in the realm of the unknown (74).
The histologic features are not specific and include focal parakeratosis, varying degrees of epidermal spongiosis with exocytosis, and a mild perivascular lymphohistiocytic infiltrate. Spongiotic vesicles, when present, contain lymphocytes and Langerhans cells.
The cutaneous eruption generally lasts for about 3 to 4 weeks, and relapses are not reported.
Lichen Sclerosus
Lichen sclerosus is generally a skin disease of adults of unknown etiology that can be seen in children (75). The majority of the affected children have involvement of the anogenital area by ivory-colored flattened papules and plaques. Human papillomavirus (HPV) has been shown to be present in some pediatric cases of lichen sclerosus (76), although the exact significance of this finding and the risk of squamous cell carcinoma in pediatric-onset lichen sclerosus are undefined (77). The clinical and histopathologic findings are essentially similar to those seen in adults. The histologic features include hyperkeratosis, epidermal atrophy, and a zone of papillary dermal sclerosis, beneath which there may be a band of lymphocytes (Figure 26-23). There is
some morphologic overlap with morphea. Lichen sclerosus in childhood generally has a better prognosis, with spontaneous resolution occurring in up to 60% of the affected girls before puberty.
some morphologic overlap with morphea. Lichen sclerosus in childhood generally has a better prognosis, with spontaneous resolution occurring in up to 60% of the affected girls before puberty.
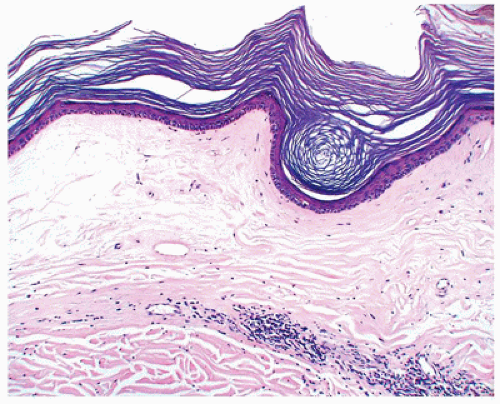 FIGURE 26-23 • Lichen sclerosus showing atrophy of the epidermis with zone of sclerosis underneath, which is a band of inflammatory cells. (Hematoxylin and eosin stain, original magnification ×200.) |
INFECTIOUS DISEASES
Bacterial Infections
Bacterial infections of skin are a common cause for pediatric outpatient visits. It may be primary or a complication of an underlying skin disease. Occasionally, skin involvement may be a manifestation of a systemic infection. Only the more common bacterial infections that often affect children are discussed.
Impetigo
Impetigo is the most common bacterial infection of the skin seen in children. Two clinical forms are recognized: nonbullous and bullous forms.
Nonbullous Impetigo (Impetigo Contagiosa)
Nonbullous impetigo or the crusted form of impetigo accounts for the majority of cases. It was historically often caused by group A β-hemolytic streptococci but now appears to be more commonly caused by Staphylococcus aureus. It is characterized by highly infectious 1- to 2-mm vesiculopustular lesions that quickly rupture to be covered by heavy yellow crusts. Lesions may involve any part of the body but occur most frequently on the exposed parts of the body such as face, neck, and extremities.
Histologic sections from a vesiculopustule show a subcorneal pustule, which may contain gram-positive cocci (Figure 26-24). Sections of the crusted lesions show a neutrophilic scale crust covering the epidermis. Impetigo contagiosa may be superimposed on pre-existing skin diseases such as atopic dermatitis (78). Complete resolution of the lesions, either spontaneously or with treatment with antibiotics, occurs in most cases. Acute glomerulonephritis, a wellrecognized sequela in a small percentage of patients, appears to be decreasing in incidence partly due to changing patterns in the infecting agents.
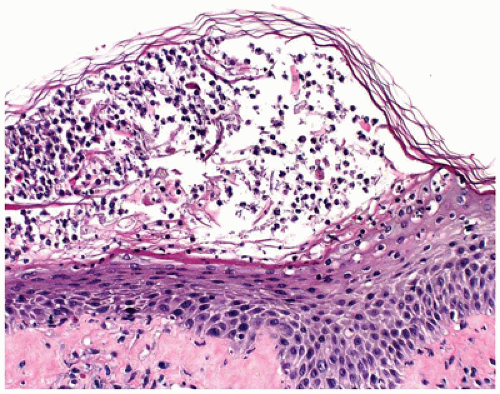 FIGURE 26-24 • Impetigo with subcorneal pustule. Gram stain may show gram-positive cocci. (Hematoxylin and eosin stain, original magnification ×200.) |
Bullous Impetigo
Bullous impetigo, almost always caused by S. aureus, generally affects newborn infants and children and can be thought of as a localized form of staphylococcal scalded skin syndrome (SSSS), caused by the same exfoliative toxins. It presents with small vesicles that may progress to flaccid bullae of more than 1 cm, with no associated erythema. The bullae are filled with clear fluid.
Histologic sections of the bullae show a cleavage plane in the uppermost part of the epidermis at or below the level of the granular layer, similar to the findings in SSSS. The underlying dermis shows a perivascular neutrophilic infiltrate that may also involve the epidermis. Unlike that in impetigo contagiosa, the bullous cavity contains few or no inflammatory cells.
When impetigo appears to be rapidly spreading, prompt treatment with systemic antibiotics avoids the risk of worsening infection or hospitalization (79). Although skin infections due to methicillin-resistant S. aureus (MRSA) are still relatively uncommon in children, given the evolving epidemiology, skin swabs should be cultured and sensitivity tests performed (80).
Staphylococcal Scalded Skin Syndrome
SSSS is a generalized blistering disease seen most often in neonates and children younger than 5 years of age. Like bullous impetigo, this disease is usually caused by epidermolytic toxin-producing S. aureus (81). The pathogen cannot be isolated from the lesions of SSSS; instead, a distant source of staphylococcal infection in the form of purulent pharyngitis, conjunctivitis, rhinitis, or umbilical infection may be present. Exfoliative toxins, ETA and ETB, produced by S. aureus target desmoglein-1, a cell-to-cell adhesion molecule found in the desmosomes of superficial epidermis, and cause the cleavage in the superficial granular layer of the epidermis, typical of SSSS (82). The clinical manifestations appear to depend on serotypes of the exfoliative toxins with ETA associated with bullous impetigo and ETB with generalized SSSS (83).
SSSS is characterized by an abrupt onset of fever and diffuse erythema that evolves into large flaccid sterile bullae filled with clear fluid. Within a short time, the bullae rupture and large sheets of epidermis peel off, giving the typical scalded appearance. The scaly desquamation resolves within 3 to 5 days without scarring.
Histologic findings are identical to those seen in bullous impetigo, with the cleavage plane at or below the granular layer. However, in contrast to bullous impetigo, the superficial dermis in SSSS is usually free of inflammatory cells (Figure 26-25). Despite the clinical similarities, SSSS can be easily distinguished from TEN, a potentially fatal skin loss disorder, based on the histologic finding of full-thickness
epidermal necrosis in the latter. In addition, mucosal involvement, often seen in TEN, is lacking in SSSS. Treatment of SSSS is directed at eradicating the nidus of staphylococcus infection and management of fluids and electrolytes with complete recovery within 2 weeks expected in most pediatric patients. Fatalities are generally related to sepsis from the primary source.
epidermal necrosis in the latter. In addition, mucosal involvement, often seen in TEN, is lacking in SSSS. Treatment of SSSS is directed at eradicating the nidus of staphylococcus infection and management of fluids and electrolytes with complete recovery within 2 weeks expected in most pediatric patients. Fatalities are generally related to sepsis from the primary source.
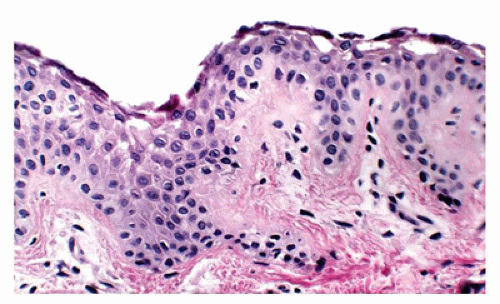 FIGURE 26-25 • Staphylococcus scalded skin syndrome showing intraepidermal clefting at the level of granular zone with minimal to absent inflammation is characteristic because the lesion is caused by toxin. (Hematoxylin and eosin stain, original magnification ×200.)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|