Fig. 27.1 Acetylcholine (ACh) at the neuromuscular junction. (A) The released ACh acts upon postsynaptic nicotinic N2 receptors on the motor endplate, opening cation channels and an influx of Na+ occurs, resulting in depolarisation. (B) At rest, insignificant amounts of ACh are released, and miniature endplate potentials generated are insufficient to reach the threshold potential to cause a propagated action potential. If sufficient ACh is released, an action potential is propagated, causing muscle contraction. Non-depolarising muscle relaxants prevent the generation of the action potential by blocking N2 receptors. AChE, acetylcholinesterase.
The neurotransmitter at the neuromuscular junction is acetylcholine (ACh), acting at postsynaptic nicotinic N2 receptors. The processes of synthesis and release of ACh are described in Chapter 4 in relation to the general properties of neurotransmitters in the nervous system. The presynaptic nerve terminal at the neuromuscular junction contains 300 000 or more vesicles, each of which may contain up to 5000 molecules of ACh (known as a quantum). In response to an action potential, up to 500 vesicles discharge their contents into the synapse over a very short period (0.5 ms). Each N2 receptor has two binding sites for ACh, and the Na+ channel in the centre of the receptor opens when both sites are occupied (Ch. 1). This allows an influx of Na + into the muscle cell and depolarisation of the motor endplate. If depolarisation is sufficient to reach the firing threshold potential for the cell then voltage-gated Na+ channels open, full depolarisation of the muscle cell is triggered and an action potential is generated and propagated along the postsynaptic membrane. Initiation of an action potential requires the release of 50–200 quanta of ACh, which will activate 10–15% of motor endplate N2 receptors (Fig. 27.1b).
The action potential generated in the skeletal muscle cell passes along the sarcolemma into the T tubules, where Ca2+ is released from the sarcoplasmic reticulum. The increased availability of intracellular Ca2+ brings about the processes that result in muscle contraction.
The action of ACh on N2 receptors is very short-lived (about 0.5 ms) because the synaptic cleft contains large amounts of the extremely potent enzyme acetylcholinesterase (AChE), which rapidly degrades ACh (Ch. 4). An esterase called pseudocholinesterase (butyrylcholinesterase, plasma cholinesterase) in plasma hydrolyses any ACh that escapes from the synaptic cleft; it acts more slowly than AChE. Pseudocholinesterase is important pharmacologically because of its ability to metabolise several drugs with ester bonds. Tissue esterases that break down ACh are also present in many cells, notably in the liver.
Although ACh is the neurotransmitter responsible for contraction of both skeletal muscle and most smooth muscles, the basic organisation and functioning of these neuroeffector systems are very different, as shown in Table 27.1.
Table 27.1
Comparison of skeletal and smooth muscle innervation
| Property | Skeletal muscle fibre | Smooth muscle fibre |
| Nerves supplying fibre | Single | Multiple |
| Junction | Highly organised motor endplate | Simple |
| Neurotransmitter | Acetylcholine | Acetylcholine |
| Receptor subtype | Nicotinic N2 | Muscarinic (mainly M3) |
| Receptor distribution | Only at motor endplate; only one motor endplate per muscle fibre | Widely on the muscle surface |
| Effects of stimulation | Single nerve contracts the whole muscle fibre (all-or-none response) | Each nerve contracts part of muscle fibre (graded response) |
| Effect of overdose with acetylcholinesterase inhibitor | Flaccid paralysis | Spasticity |
Drugs acting at the neuromuscular junction
Acetylcholinesterase inhibitors
AChE inhibitors block the breakdown of ACh following its release in neuronal synapses and at neuroeffector junctions. The mechanisms of action of different types of AChE inhibitor are described in Chapter 4; they are non-selective and prolong the availability and actions of ACh at all its receptors (nicotinic N1 and N2 and muscarinic). Full details of AChE inhibitors in the treatment of myasthenia gravis are given in Chapter 28.
Inhibitors of acetylcholine release
Botulinum toxin from the anaerobic bacillus Clostridium botulinum decreases the release of ACh from vesicles. It binds selectively to cholinergic nerve terminals and, after internalisation via a cell membrane vesicle, is released into the cell cytoplasm. The toxin cleaves cytoplasmic proteins (SNARE proteins) on the cell membrane that are essential for docking and fusion of vesicles with the neuronal membrane, and this inhibits neurotransmitter release. This chemical denervation stimulates collateral axon growth which eventually results in the formation of a new neuromuscular junction. Botulinum toxin is extremely dangerous, as evidenced by the consequences of botulinum poisoning, but it also has clinical roles when injected locally. Two toxin serotypes, A and B, are used in clinical practice as botulinum toxin–haemagglutinin complex. Injection into skeletal muscles produces local muscle paralysis for many weeks until new nerve terminals develop. Botulinum toxin is used to treat involuntary movements such as blepharospasm (spasm of the eyelids) or torticollis (wry-neck) and to relieve spasticity (Ch. 24). It is also used by local injection to reduce excessive sweating, because it inhibits ACh release at sweat glands, and is occasionally injected into facial muscles for the prophylaxis of migraine (Ch. 26). Botulinum toxin is increasingly being used for cosmetic reasons to temporarily remove frown lines and wrinkles.
Antagonists/blockers at the neuromuscular junction
Skeletal muscle relaxation is achieved by drugs that specifically and reversibly block the actions of ACh on nicotinic N2 receptors at the neuromuscular junction; they do not affect autonomic nervous system function (i.e. the actions of ACh on nicotinic N1 receptors in ganglia or on muscarinic receptors at postganglionic nerve endings). Drugs that block the neuromuscular junction almost all resemble ACh in that they have a quaternary ammonium (N+) group that binds strongly to the anionic site of the nicotinic N2 receptor (Fig. 27.2).
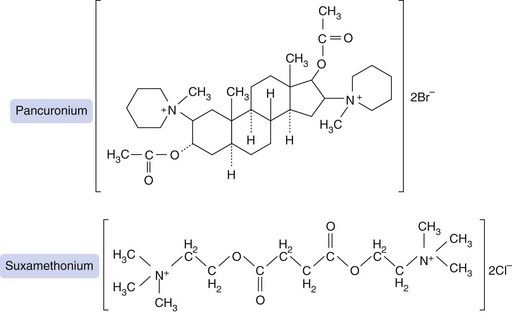
Fig. 27.2 Structures of pancuronium, a non-depolarising blocking drug, and suxamethonium (succinylcholine), a depolarising blocker. Suxamethonium resembles two molecules of acetylcholine, each with a quaternary ammonium (N+) group, linked back-to-back. Pancuronium was designed to have a similar spatial arrangement of quaternary nitrogens, but held rigidly in place by a steroid bridge that is resistant to pseudocholinesterase.
A neuromuscular blocker must occupy more than 75% of postsynaptic N2 receptors before it can start to produce neuromuscular blockade. The potency of a neuromuscular blocker is measured by the ED95, which is the dose required to produce 95% depression of muscular twitch (Table 27.2). About twice this dose is required for muscle relaxation adequate to permit tracheal intubation. The laryngeal muscles are more rapidly paralysed than other skeletal muscle groups, but the effect is often of shorter duration. This may reflect either the higher blood flow to this muscle, or the greater density of N2 receptors.
Table 27.2
Properties of some neuromuscular junction-blocking drugs
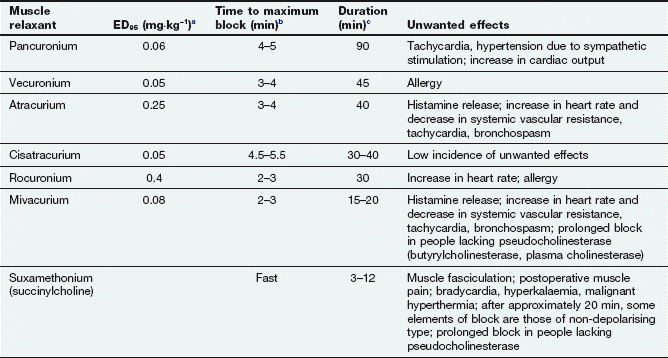
aED95 is the effective dose required to suppress muscle twitch by 95%.
bTime to maximum block following administration of the dose used for intubation (double the ED95).
cTime taken to recover to 25% of the original twitch height after an intubation dose (double the ED95).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


