The Multiple Organ Dysfunction Syndrome: Prevention and Clinical Management
John C. Marshall
The multiple organ dysfunction syndrome (MODS) is the final common pathway to death for most critically ill patients who die within the contemporary health care system. It is a process that is all too familiar to the surgeon, often arising as a complication of treatment, and heralding an inexorable trajectory toward major morbidity or death. It is, moreover, an intimidatingly complex process that integrates a rapidly expanding and unfamiliar biology with some of the most sophisticated supportive technologies in health care, and that evolves at the interface between heroic care and inappropriate meddling in the process of dying. MODS is both a complication to be prevented and a reflection of the successes and limitations of contemporary critical care.
Critical illness is fundamentally different from other conditions that the surgeon manages. Unlike diseases such as acute appendicitis or subarachnoid hemorrhage, it lacks a discrete and definable locus for intervention, through a broad spectrum of diseases may be its antecedents. Unlike cancer or localized inflammatory conditions, it lacks defining histological features that permit the definition of a discrete pathologic state, though disseminated processes such as inflammation, thrombosis, or ischemia are commonly present. Most importantly, however, the unique feature of critical illness that defines it, and makes its management so challenging, is the dependence of the phenotype and course on the very clinical interventions used to sustain life in a patient who would otherwise have died a rapid death.
The construct of MODS embodies these features, and is at once a description of the clinical course of a patient over time, a consequence of tissue injury and the host response to that injury, and a metaphor for the changing processes of ICU care.
Multiple Organ Dysfunction Syndrome and the Evolution of Intensive Care
The capacity to resuscitate the mortally injured or ill patient, and to sustain life through the provision of exogenous life support measures, is a product of insights garnered during the twentieth century, often, ironically, during the conduct of war. At the time of the World War I, for example, abdominal wounds carried such a high mortality rate that surgical intervention was deemed futile and inappropriate. Pioneering studies by Blalock and others less than 100 years ago inform our current understanding of the role that reduced intravascular volume plays in the pathogenesis of shock. The very word “shock” reflects the mistaken belief prevalent at the time that the syndrome was a psychological state reflecting fear of impending death, rather than a readily correctible circulating fluid deficit. Recognition of the importance of intravascular fluid loss enabled crystalloid resuscitation and blood transfusion, and salvaged patients who in an earlier era would have died.
Dialytic techniques to support the failing kidney were developed in the aftermath of World War II, while positive pressure mechanical ventilation was an innovation of polio epidemics of the early 1950s. Techniques for accessing and monitoring the central venous system were also developed in the 1950s, and enabled more precise characterization of the hemodynamic alterations of shock. These innovations set the stage for the development of the intensive care unit (ICU) as a geographic locale where life support technologies could be administered; they fundamentally altered the processes through which the multiply traumatized or critically ill patient proceeded toward recovery or death.
The development of novel life support technologies also spawned the emergence of new clinical syndromes. Acute renal failure following crush injuries or multiple traumas was first described in the 1940s and 1950s; its description only became possible as techniques of renal dialysis enabled patients with acute renal failure to live more than several days. Similarly, the acute respiratory distress syndrome (ARDS) only became a relevant concept when it was possible to survive acute respiratory failure through the intervention of mechanical ventilation, while septic shock is a disorder that arose with the capacity of medical technology to support the profound cardiovascular derangements of systemic inflammation.
With the widespread establishment of ICUs in large hospitals during the 1960s, a spectrum of new clinical disorders emerged—stress-induced upper gastrointestinal bleeding, disseminated intravascular coagulation, gram-negative septicemia, ICU jaundice, and ARDS to list the more prominent. The concept that these were varied manifestations of a common process was first articulated by Arthur Baue, who, in 1975, suggested that each was a manifestation of a process that was better understood as multiple systems failure. Epidemiologic studies of this new and common ICU syndrome revealed a number of discrete risk factors, all of which reflect the activation of an innate immune response to a potentially life-threatening insult (Table 1).
Terminology and Definitions
The clinical syndrome of organ failure in the critically ill patient has many synonyms—multiple organ failure, multiple systems organ failure, and remote organ failure, to name a few. A consensus conference held in 1991 proposed the terminology “the Multiple
Organ Dysfunction Syndrome (MODS)” to emphasize several key aspects of the process. First, organ system insufficiency in MODS is appropriately seen as dysfunction rather than failure, for it is variable in severity and potentially reversible. Second, the process is a systemic one, reflected in the involvement of more than one organ system, hence the terminology “multiple organ.” Finally, although the mechanisms of the process are incompletely understood, it arises through the activation of a systemic inflammatory response, and so can be considered a syndrome—a descriptive term for a disorder that is defined on the basis of clinical manifestations that are believed to have common underlying pathologic mechanisms.
Organ Dysfunction Syndrome (MODS)” to emphasize several key aspects of the process. First, organ system insufficiency in MODS is appropriately seen as dysfunction rather than failure, for it is variable in severity and potentially reversible. Second, the process is a systemic one, reflected in the involvement of more than one organ system, hence the terminology “multiple organ.” Finally, although the mechanisms of the process are incompletely understood, it arises through the activation of a systemic inflammatory response, and so can be considered a syndrome—a descriptive term for a disorder that is defined on the basis of clinical manifestations that are believed to have common underlying pathologic mechanisms.
Table 1 Risk Factors for the Development of MODS | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The same consensus conference also sought to clarify terminology related to the complex interplay of host and microbial factors that shapes the clinical syndrome of sepsis in the critically ill patient. Infection is the invasion of normally sterile host tissues by viable microorganisms. This process may or may not evoke an inflammatory response that can occur in the local environment of the infectious challenge (e.g., a surgical wound infection) or may be a disseminated systemic process; the latter defines sepsis—the systemic host inflammatory response to invasive infection. Because the response may be variable in its expression and inflammation is a normal adaptive response to infection, sepsis was further characterized as severe sepsis (sepsis in association with organ dysfunction) and septic shock (sepsis resulting in hemodynamic compromise). But it is also evident that a systemic host response that is clinically indistinguishable from sepsis can also be evoked by noninfectious stimuli; the terminology systemic inflammatory response syndrome (SIRS) was proposed to describe the clinical response independent of its cause.
The consensus conference terminology has been widely adopted, and the relationship between these differing entities illustrated schematically (Fig. 1). While the concepts are useful, they have inherent limitations. Most importantly, the classic SIRS criteria—tachycardia, tachypnea, hyper- or hypothermia, and leukocytosis or leukopenia—are nonspecific, and readily evoked by noninflammatory stimuli such as vigorous exercise. They were proposed not on the basis of epidemiologic data but on the basis of an arbitrary consensus process. Subsequent studies have shown that they are present in the majority of patients in an ICU, and equally, that serious infection may be present when SIRS criteria are not met. A more recent consensus conference has proposed additional clinical criteria that are characteristic of systemic inflammation, but expansion of the definition only serves to reduce its specificity. We currently lack a well-characterized clinical syndrome of systemic inflammation, though the classic cardinal signs of inflammation defined by Galen and Celsus two millennia in the past provide a framework for thinking of the clinical syndrome, and incorporate MODS as the systemic counterpart of the local loss of function that is characteristic of acute inflammation (Table 2).
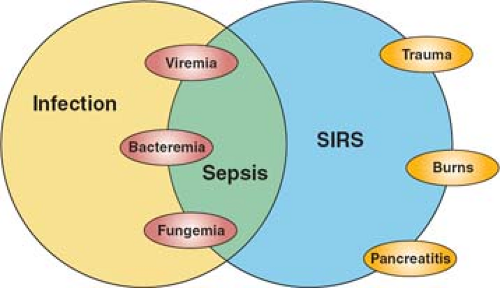 Fig. 1. The relationship between sepsis and infection. Infection denotes the microbial process—invasion of normally sterile tissues by microorganisms, while sepsis describes the systemic host response that results from infection. The response can occur following noninfectious insults, and so the term systemic inflammatory response syndrome (SIRS) describes that response independent of its cause. (From Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. ACCP/SCCM CONSENSUS CONFERENCE. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992;101:1644–55.) |
The Pathogenesis of Multiple Organ Dysfunction Syndrome
The Cellular Biology of Inflammation
Multicellular organisms have lived in intimate proximity with the microbial world for more than a billion years. The very existence of complex organisms is a consequence of the fact that primitive unicellular species were parasitized by proteobacteria that became the mitochondria of animals and the chloroplasts of plants, and that enabled cells to generate energy and to develop specialized functions as part of a larger organism. But microorganisms in the environment also pose a potential threat, and the emergence of multicellularity occurred in parallel with the evolution of elegant mechanisms that serve to recognize danger and to respond effectively in such a way that host homeostasis is restored. These mechanisms are varied and intricate, and include specialized immune cells, the coagulation cascade, and literally hundreds of host-derived molecules that modify cellular physiology and support a coordinated response to danger. One of the most important of these systems—based on a family of genetically encoded receptors called toll-like receptors (TLRs)—is described in greater detail (Fig. 2).
Table 2 Cardinal Manifestations of Local and Systemic Inflammation | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TLRs represent a family of 11 proteins expressed on the surface of cells of the innate immune system as well as on nonimmune tissues such as epithelial cells. The ligands for TLRs are molecules, whose presence in the microenvironment of the cell signifies the possibility of danger; as a class, these ligands have been termed pathogen-associated molecular patterns (PAMPs), or more accurately damage-associated molecular patterns (DAMPs). For example, endotoxin—a lipopolysaccharide that is the major constituent of the cell wall of gram-negative bacteria—binds to and activates TLR4, while TLR2 is activated by molecules such as peptidoglycan and lipoteichoic acid that are found in the cell wall of gram-positive bacteria. Viral RNA or DNA is recognized by TLR3, TLR7, and TLR8, while characteristic motifs in bacterial DNA activate TLR9. Flagellin, a protein found in the flagellae of bacteria, activates
TLR5. In contrast to the recognition molecules of the adaptive immune system—the T-cell receptor and specific antibody—TLRs are encoded in the genome, and constitutively expressed on cells; thus they are rapidly activated when their ligand is present. A degree of specificity results from the specific TLR that is engaged, but because the cellular mechanisms downstream of the receptor are similar, the response is not specific for the cause, at the level of either the cell or the whole organism.
TLR5. In contrast to the recognition molecules of the adaptive immune system—the T-cell receptor and specific antibody—TLRs are encoded in the genome, and constitutively expressed on cells; thus they are rapidly activated when their ligand is present. A degree of specificity results from the specific TLR that is engaged, but because the cellular mechanisms downstream of the receptor are similar, the response is not specific for the cause, at the level of either the cell or the whole organism.
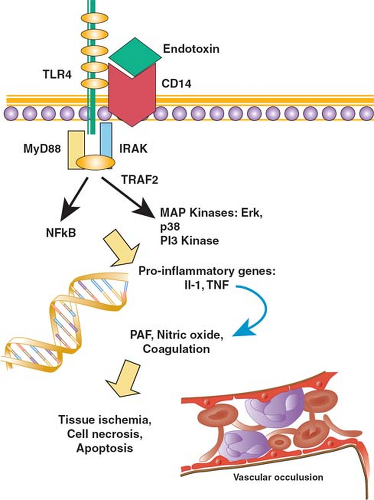 Fig. 2. A schematic representation of the cellular events that occur in response to infection or tissue injury. Engagement of a pattern recognition receptor on the surface of cells such as neutrophils and monocytes (e.g., engagement of TLR4 by endotoxin) results in the recruitment of signaling molecules to the intracellular tail of the receptor, and activates intracellular pathways that result in the expression of early proinflammatory genes such as interleukin-1β and tumor necrosis factor (TNF). These proteins, in turn, act on cells through their own specific receptors, inducing the expression of a broad variety of mediators that have been implicated in the pathogenesis of inflammation. |
TLRs are activated by both microbial molecular patterns and by certain host-based molecules whose presence outside the cell signals injury, and so danger. In addition to endotoxin, for example, TLR4 can be activated by heparan sulfate, heat shock proteins, oxidized phospholipids, and other substances released by injured host tissues. Reflecting their ancient bacterial origins, mitochondrial products released as a result of cellular injury can activate innate immune cells through interactions with TLR9. Thus, at both a clinical and a molecular level, the host response is not specific to infection, but rather is more appropriately understood as a response to danger.
The engagement of a TLR initiates a signal that is transmitted to the nucleus of the cell, where it results in the expression of some genes and in the inhibition of others. The process called signaling involves multiple cellular proteins but common biochemical principles. When the extracellular region of a TLR is engaged by its ligand, the conformation of the intracellular portion of the receptor is altered. This change results in clustering of proteins associated with the receptor, and in secondary changes to the structure of the intracellular portion of the receptor. A particularly important example of the latter is phosphorylation—the addition of a phosphate group derived from ATP to specific amino acids (in particular tyrosine, serine, and threonine) in the intracellular tail of the receptor. The addition of a phosphate group occurs through the action of enzymes called kinases, while the removal of a phosphate group is effected by phosphatases. By changing the shape and charge of the receptor tail, phosphorylation or dephosphorylation enables the receptor to recruit additional signaling molecules from the cytoplasm, and so to begin the process of transmitting the environmental signal to the nucleus to induce changed patterns of gene expression.
Transduction of a signal from the cell surface to the nucleus results from the serial activation of intracellular enzymatic cascades, for example, the mitogen-activated protein (MAP) kinase cascades and the phosphatidyl inositol-3 (PI3) kinase cascade. Cytoplasmic proteins called transcription factors are activated, and translocate to the nucleus where they bind to the promoter region of target genes, and initiate or inhibit the process of gene expression or transcription. A transcription factor called NFκB plays a particularly important role in initiating the transcription of a number of inflammatory genes.
The early response to TLR activation is the synthesis and release of early proinflammatory cytokines. Cytokines are proteins whose expression is induced in response to an immunologic stimulus, and which can, in turn, bind specific receptors and evoke a response in their target cells. Interleukin-1 (IL-1) and tumor necrosis factor (TNF) are particularly important examples of cytokines that are released early, following cellular exposure to an inflammatory stimulus. Cytokines induce a further cascade of signal transduction and new gene expression including the expression of key genes that shape the clinical phenotype of systemic inflammation. For example, cytokine-mediated upregulation of the enzyme inducible nitric oxide synthase (iNOS) in endothelial cells results in increased enzymatic activity and the
conversion of the amino acid arginine to citrulline, a process that generates a molecule of the short-lived gas, nitric oxide. Nitric oxide is a potent vasodilator, and increased local production results in vascular smooth muscle relaxation and the characteristic vasodilatation of inflammation. Increased expression of tissue factor on endothelial cells induces local activation of the coagulation cascade, and microvascular thrombosis. Cytokine-mediated injury to the tight junctions between endothelial cells permits fluid extravasation into the interstitium, producing edema.
conversion of the amino acid arginine to citrulline, a process that generates a molecule of the short-lived gas, nitric oxide. Nitric oxide is a potent vasodilator, and increased local production results in vascular smooth muscle relaxation and the characteristic vasodilatation of inflammation. Increased expression of tissue factor on endothelial cells induces local activation of the coagulation cascade, and microvascular thrombosis. Cytokine-mediated injury to the tight junctions between endothelial cells permits fluid extravasation into the interstitium, producing edema.
The forgoing description is necessarily simplified. Innate immune cells can be activated through receptors other than those of the TLR family. Endotoxin exposure in the healthy human volunteer leads to differential expression of more than 3,700 genes, and for many of these, little is known about their pathogenic role in systemic inflammation. The subsequent activation of genes that amplify, suppress, or modulate those genes expressed in response to TLR stimulation results in a bewilderingly complex cascade of cascades whose key features defy simple characterization. Nonetheless, in aggregate they support integrated responses that generate key features of systemic inflammation, and so are intimately implicated in the pathogenesis of MODS.
From Inflammation to Organ Dysfunction
Activation of a systemic inflammatory response evokes a diverse series of alterations in homeostasis at the cellular level. How these changes result in physiologic dysfunction at the level of the organ or entire organism remains speculative, although a number of models have been proposed. These are by no means mutually exclusive, and probably better conceptualized as differing perspectives on a very complex process. Moreover, the histologic changes in failing organs are variable and surprisingly bland, and in the absence of characteristic pathologic changes, a single pathogenic mechanism is difficult to discern. The mechanisms of MODS at the organ level can be thought of as arising from three overlapping processes (Fig. 3).
Direct tissue injury from ischemia and inflammation
Remote cellular responses to circulating factors released in response to tissue injury
“Second hits”—further injury resulting, for example, from products from the gut, the environment, or the adverse effects of ICU supportive care.
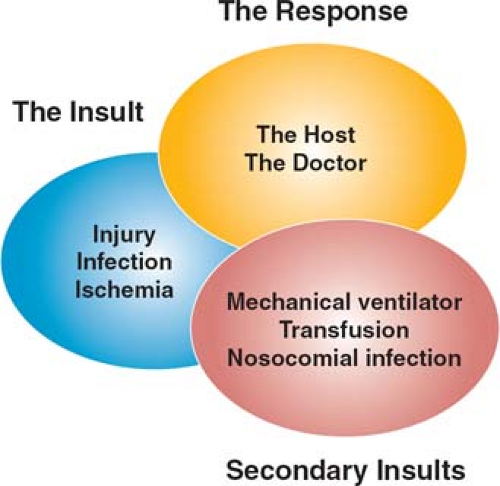 Fig. 3. The evolution of multiple organ dysfunction syndrome (MODS) occurs through the interactions of the initial insult with the inflammatory response of the host and the clinical response of the doctor, modified by subsequent insults or “second hits” that include ICU-acquired infection or the adverse consequences of interventions such as transfusion, ventilation, or drugs. |
Tissue Injury Induced by Ischemia and Inflammation
Activation of the innate immune system provides a rapid and effective defense against invading microorganisms, but at a cost of local tissue injury. Neutrophils release reactive oxygen intermediates and a number of proteases that kill bacteria but also injure host cells, while monocytes and macrophages release an extensive repertoire of proinflammatory mediators following the recognition of bacterial DAMPS; these agents alter cellular metabolism and blood flow locally, and induce activation of the coagulation cascade. This sequence of cellular responses is responsible for the characteristic changes seen in the organs of the patient with MODS.
The early stages of the ARDS, for example, are characterized by interstitial edema, vascular thrombosis, and massive neutrophilic infiltration (Fig. 4). This response can be precipitated by a local insult in the lung, but can also result from a remote insult such as intestinal ischemia and reperfusion that triggers neutrophil activation, cytokine release from macrophages, and the expression of adhesion molecules on the pulmonary vascular endothelium. Activated neutrophils bind to adhesion molecules on endothelial cells in the lung, and then extravasate into the pulmonary interstitium. Damage to the epithelial tight junctions results in increased extravasation of protein-rich fluid into the interstitial space, producing permeability edema; oxygenation is impaired since alveolar oxygen must diffuse across a greater distance to reach adjacent capillaries. Cytokine-mediated expression of tissue factor on pulmonary endothelial cells activates the coagulation cascade, producing focal areas of microvascular thrombosis. The generation of fibrin as the end product of the coagulation cascade initiates the process of tissue repair, but this fibrosis also creates an additional barrier to gas exchange in the lung, a characteristic feature of the late stages of ARDS.
Multiple factors contribute to cellular hypoxia during an inflammatory response (Fig. 5). The unloading of oxygen from the red blood cell to the tissue occurs by passive diffusion down a gradient dependent on the difference in oxygen tension between the red cell and the tissues. This process occurs in the microvasculature, where transit is slowest, and the distance between blood cells and cells of the adjacent tissues is minimal. Interstitial edema increases this distance, with the result that the diffusion gradient is less steep, and less oxygen is unloaded. Reduced vascular resistance results in both a faster rate of flow through the microvasculature and shunting of oxygenated blood, both of which limit the offloading of oxygen. The formation of anatomic shunts is further exacerbated as individual capillaries become occluded by adherent white cells, red cells, and platelet thrombi resulting from activation of the coagulation cascade. The net result is that although blood entering the capillary bed is adequately oxygenated, it unloads much less of its carried oxygen, and so oxygenation of tissues is
impaired. At the same time, venous blood returning to the heart carries more than the normal content of oxygen (normal values are about 70% saturation), and the mixed venous oxygen saturation is characteristically increased. Finally, there is some evidence that oxygen utilization by the tissues is impaired in sepsis, a state that has been termed “cytopathic hypoxia.”
impaired. At the same time, venous blood returning to the heart carries more than the normal content of oxygen (normal values are about 70% saturation), and the mixed venous oxygen saturation is characteristically increased. Finally, there is some evidence that oxygen utilization by the tissues is impaired in sepsis, a state that has been termed “cytopathic hypoxia.”
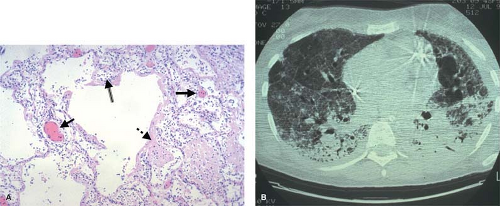 Fig. 4. A: The histologic features of acute respiratory distress syndrome (ARDS) reflect several concomitant pathologic processes including the infiltration of neutrophils (double-lined arrow), intravascular thrombosis (solid arrows) and fibrin deposition (dashed arrow). B: Although ARDS appears as diffuse fluffy infiltrates on a chest x-ray, a CT scan reveals the process to be inhomogeneous, and reflects the role of clinical factors in its development—atelectasis in dependent lung zones resulting from nursing the patient in a supine position and cystic changes in the antidependent regions reflecting injury caused by overdistention of the lung by the mechanical ventilator. |
In summary, tissue oxygenation in the patient with MODS is impaired for multiple reasons, and the function of the hypoxic cell is altered. Prior to resuscitation, intravascular volume loss and reduced systemic vascular resistance combine to render tissues ischemic. With resuscitation, products from ischemic tissues activate the inflammatory response, with the result that endothelial adhesion molecule expression is increased, and activated neutrophils migrate into tissues where their products induce further injury. The local inflammatory response results in increased capillary permeability and tissue edema, as well as endovascular activation of coagulation, further compromising oxygen uptake at the tissue level, and creating a vicious cycle of local tissue injury.
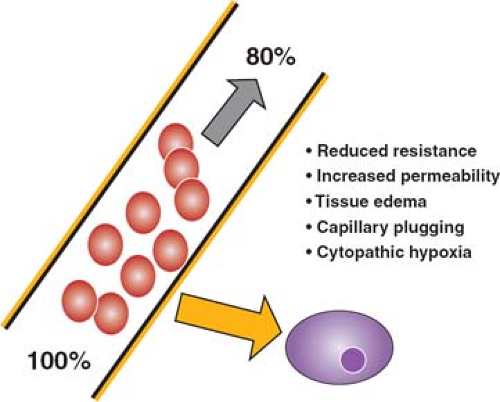 Fig. 5. Under normal circumstances, blood leaving the lungs is 100% saturated with oxygen; as it passes through the capillary bed, about 30% of that carried oxygen diffuses into the adjacent tissues, so that the blood returning to the heart is only 70% saturated. In sepsis, the amount of oxygen unloaded is less, so that the returning venous blood has an oxygen saturation of 80% or even higher. Factors contributing to the failure of off-loading of oxygen include reduced vascular resistance resulting in faster transit of blood, with less time for release of oxygen, increased endothelial permeability and tissue edema resulting in a reduced gradient for oxygen diffusion, capillary plugging leading to shunting, and impaired oxygen uptake at the cell membrane. |
Remote Cellular Responses to Circulating Inflammatory Products
Cellular dysfunction can be a direct consequence of cellular hypoxia as discussed above. It can also result from cellular injury or death, either by necrosis or by the programmed cell death or apoptosis of the cell.
Profound cellular hypoxia or the products of activated neutrophils can induce structural damage to the cell with the result that membrane integrity is lost, and intracellular constituents leak into the extracellular environment. This process is known as necrosis; as discussed above, constituents of the cell, which are normally intracellular, function as DAMPs in the extracellular space, and activate local inflammation by engaging TLRs. Necrosis evokes inflammation, and the resulting inflammatory response results
in further cellular necrosis, amplifying the injury.
in further cellular necrosis, amplifying the injury.
Apoptosis is a noninflammatory mode of physiologic cell death that plays a fundamental role in normal homeostasis during embryogenesis, cell turnover, and immune surveillance for infected or transformed cells. For example, in health, neutrophils are the shortest-lived cell of the human body, constitutively dying by apoptosis within hours of their release from bone marrow. Each day, in excess of ten billion neutrophils are released from bone marrow stores and a comparable number die by apoptosis. Apoptosis not only removes aged, transformed, or infected cells, but the uptake of apoptotic cell remnants by the reticuloendothelial system induces the expression of genes that support the resolution of inflammation and the processes of tissue repair. Apoptosis can be initiated by proteins that engage death receptors on the surface of the target cell. The receptor for TNF is such a receptor, and the tumor-killing properties of this cytokine derive from its ability to evoke apoptosis in the transformed cell.
Cellular apoptosis during MODS may be either excessive or inadequate. Increased rates of apoptosis are evident in lymphocyte populations (accounting for the characteristic lymphopenia of critical illness) and in epithelial cells, particularly those of the gut and kidney. In contrast, the apoptosis of neutrophils is inhibited, with the result that activated and potentially injurious neutrophils persist in the circulation, and the normal stimulus to tissue repair that results from the phagocytosis of apoptotic cells is dampened.
“Second Hits” Inducing Further Tissue Injury
The initial disease process leading to life-threatening critical illness is usually the dominant insult experienced by the patient; however, the process of resuscitation and support in the ICU creates a series of secondary insults that can modify or perpetuate the response to the initial problem. This notion is embodied in the concept of a “second hit.” Its sources are many.
Nosocomial infection is an important second hit in the critically ill patient. ICU-acquired infections differ in important respects from the infections that characteristically lead to ICU admission. The infecting flora includes organisms such as coagulase-negative Staphylococci, Enterococci, Pseudomonas, and Candida—species that have low intrinsic virulence, and that are uncommon in community-acquired infections. These organisms, however, share features that account for their predominance in infections in the critically ill patient. They grow in biofilms on synthetic surfaces such as endotracheal tubes and intravascular and urinary catheters, and established colonies on these devices are difficult to eradicate. They also are the predominant species that colonize the proximal gastrointestinal tract of the critically ill patient, and each is capable of translocating in an intact and viable form across an intact gut mucosa. Finally, they are resistant to common first-line antibiotics used in an ICU, and are capable of increasing their intrinsic virulence in an acutely ill host. Other common organisms causing nosocomial ICU-acquired infection emerge as pathogens under a combination of antibiotic resistance and person-to-person transmission (methicillin-resistant Staphylococcus aureus or Clostridium difficile) or because they persist and thrive in the hospital water supply (Acinetobacter).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


