no response to antibiotic therapy or a failure to regress after 6 weeks without identification of an infectious etiology (6).
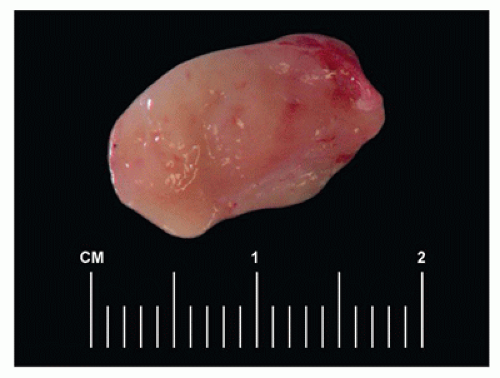 FIGURE 23-1 • Gross examination of a reactive lymph node in a child shows tan vaguely nodular cut surface. |
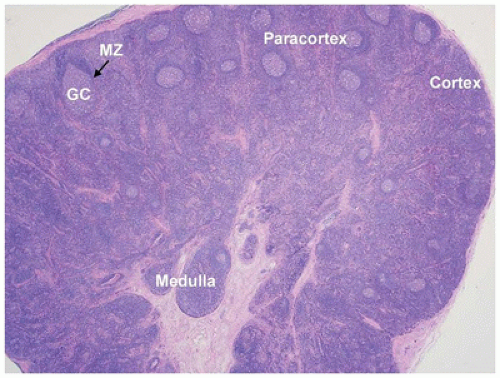 FIGURE 23-2 • The lymph node consists of three recognizable compartments: cortex (B-cell-rich zone), paracortex (T-cell-rich zone), and medulla. The secondary follicles located in the cortical areas show germinal center (GC) and well-demarcated mantle zone (MZ). (Hematoxylin and eosin stain 1.25× magnification.) |
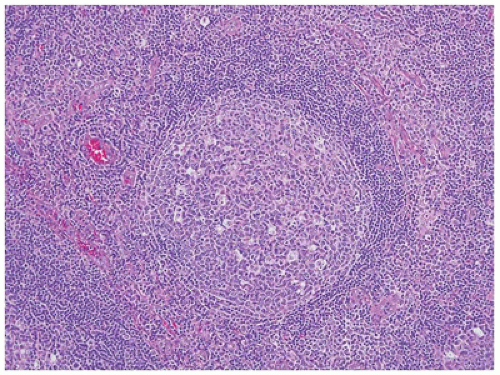 FIGURE 23-3 • The secondary follicles show a reactive germinal center, surrounded by a well-demarcated mantle zone. Polarization of the benign germinal center reflects the segregation of centrocytes to the light zone and mitotically active centroblasts to the dark zone. (Hematoxylin and eosin stain 10× magnification.) |
generated. In the appropriate clinical setting, some karyotypic abnormalities may be pathognomonic for certain types of malignancies (Table 23-3) and can therefore be used for diagnostic/classification purposes (8,9). In other settings, especially precursor B-cell lymphoblastic lymphoma/leukemia, the results of cytogenetic studies can also be used for prognostic purposes (10). The most common karyotypic changes related to lymphoproliferative disorders in children include translocations of immunoglobulin and T-cell receptor loci, which are frequently paired with loci involved in normal development and hematopoiesis (11,12). A major advance in diagnostic cytogenetics is the widespread availability of fluorescent in situ hybridization (FISH) studies for these translocations. Two major advantages of FISH techniques over routine cytogenetics are (a) rapid turnaround (<24 hours) and (b) ability to utilize fixed tissues including touch preparations, cytospin preparations, or sections of paraffin-embedded tissues (13,14).
TABLE 23-1 APPROACH TO DIAGNOSIS AT THE TIME OF BIOPSY | |
|---|---|
|

TABLE 23-2 MARKERS USEFUL IN DIAGNOSIS OF HEMATOLYMPHOID NEOPLASMS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 23-3 KARYOTYPIC AND GENETIC CHANGES ASSOCIATED WITH NON-HODGKIN LYMPHOMA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 23-4 REACTIVE LYMPHADENOPATHY IN CHILDREN | |
|---|---|
|
of the paucity of lymphocytes. The morphologic features associated with persistent generalized lymphadenopathy (i.e., large and irregularly shaped follicles, follicular lysis, follicular involution) are distinctive but not specific for HIV infection, as they have been seen in 5% to 10% of otherwise entirely unremarkable lymph nodes obtained as part of carcinoma staging before the beginning of the AIDS era (20).
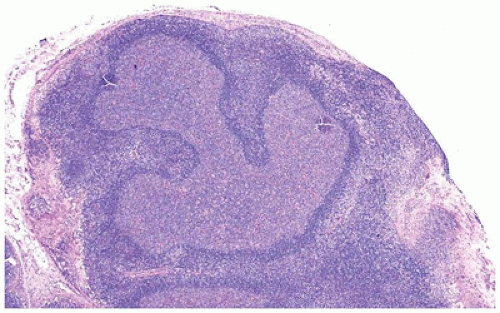 FIGURE 23-4 • Serpentine follicular center in a patient with HIV infection and persistent generalized lymph adenopathy. (Hematoxylin and eosin stain 4× magnification.) |
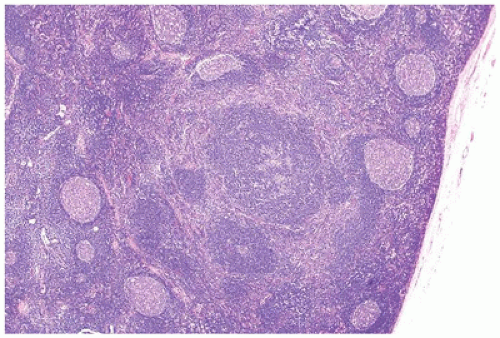 FIGURE 23-5 • Progressively transformed germinal center (with disrupted follicle infiltrated by small mantle zone lymphocytes) in a background of follicular hyperplasia. (Hematoxylin and eosin stain 4× magnification.) |
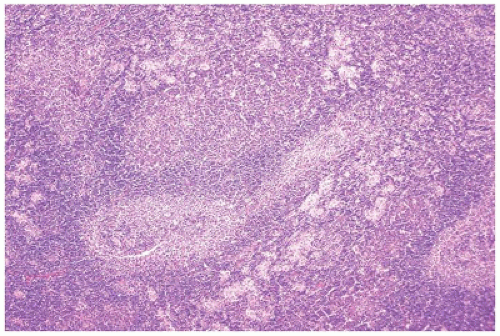 FIGURE 23-6 • Low power of toxoplasmosis demonstrating the triad of hyperplastic follicles, monocytoid B-cell proliferation, and histiocytic aggregates encroaching of follicles. (Hematoxylin and eosin stain 4× magnification.) |
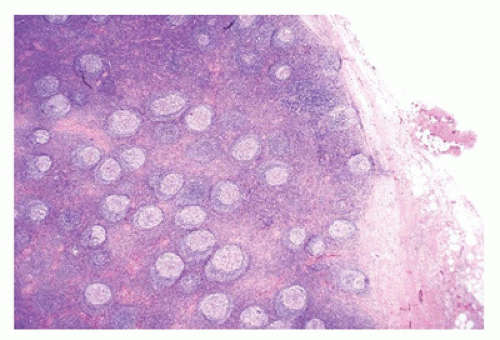 FIGURE 23-7 • Low power of hyaline vascular Castleman disease with “bag of marbles”: small uniform follicles evenly dispersed throughout the cortex and the medulla. (Hematoxylin and eosin stain 4× magnification.) |
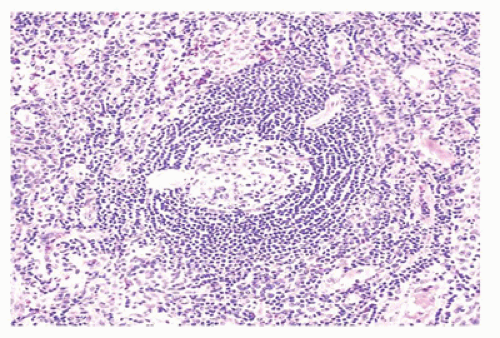 FIGURE 23-8 • The mantle zone has a laminated or “onionskin” appearance in the hyaline vascular type of Castleman disease. Note the radially penetrating vessel. (Hematoxylin and eosin stain 10× magnification.) |
Normal landmarks—germinal centers and subcapsular and paratrabecular sinuses—are generally present but may be compressed or distorted by the immunoblastic proliferation. In very early cases of EBV infection, monocytoid B-cell proliferation may be prominent (36). Staining with CD20 and CD3 highlights the presence of a mixture of interfollicular B and T immunoblasts, respectively, and a polytypic pattern of light-chain expression is always seen. The Reed-Sternberg-like cells are characteristically CD20+ and CD15- and show variable reactivity for the activation antigen CD30 as well as markers of EBV infection such as latent membrane protein (LMP1) or EBV-encoded RNA transcripts (EBER) (37). Difficult cases may exhibit sheet-like arrays of immunoblasts, a brisk mitotic rate, or extensive necrosis and may closely mimic large-cell lymphoma. In such cases, examination of the peripheral blood smear for atypical lymphocytes, viral serology, and immunohistochemistry to better define architectural preservation and establish the presence of EBV is helpful.
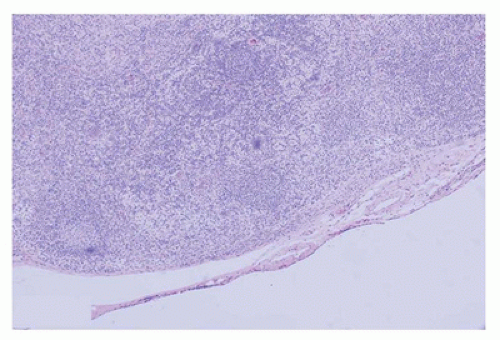 FIGURE 23-9 • The immunoblastic proliferation in acute infectious mononucleosis localizes to the paracortex and may compress or distort residual germinal centers. (Hematoxylin and eosin stain 4× magnification.) |
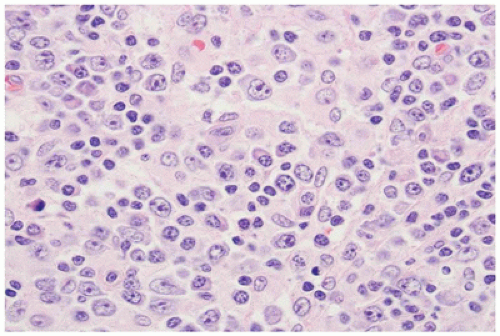 FIGURE 23-10 • Small, intermediate, and large cells fill the paracortex in acute infectious mononucleosis, often with a predominance of immunoblasts. (Hematoxylin and eosin stain 40× magnification.) |
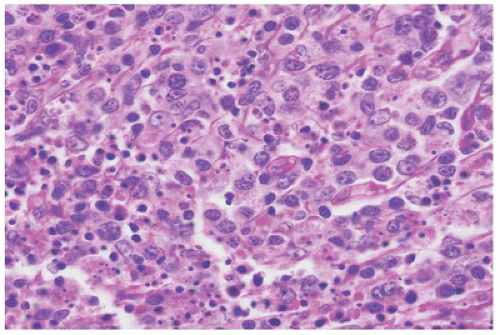 FIGURE 23-11 • The border of necrosis shows apoptotic debris admixed with histiocytes, small lymphocytes, plasmacytoid dendritic cells, and immunoblasts in Kikuchi disease. (Hematoxylin and eosin stain 40× magnification.) |
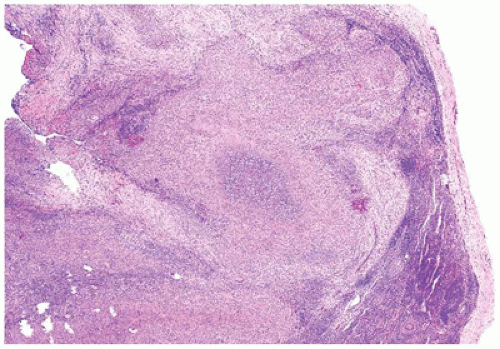 FIGURE 23-12 • The microabscesses in cat-scratch disease have a serpiginous or stellate contour. (Hematoxylin and eosin stain 4× magnification.) |
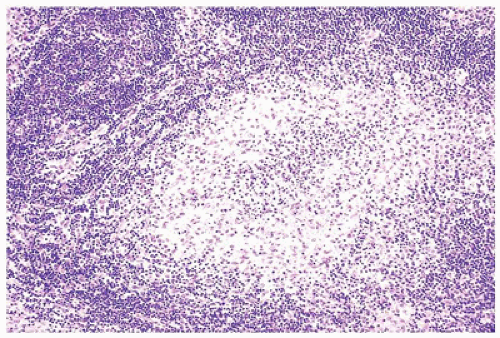 FIGURE 23-13 • In well-developed cases, the abscesses of cat-scratch disease have a broad rim of palisading histiocytes surrounding abundant neutrophils forming a so-called pyogranuloma. (Hematoxylin and eosin stain 10× magnification.) |
the MPT64 mycobacterial antigen (more specific for the Mycobacterium tuberculosis complex) (66,67). The remainder of cases may be diagnosed via one of several polymerase chain reaction-based techniques (68,69,70,71,72) or microbiologic culture. Most of these techniques, except for culture, have the advantage of being applicable to fresh as well as paraffin-embedded tissue and may be performed using cytology specimens, core needle biopsies, or whole lymph node biopsies. Other causes of caseating and noncaseating granulomatous lymphadenitis include nonmycobacterial infections and neoplastic disease, including peripheral T-cell lymphoma, NLPHL, and classical Hodgkin lymphoma (CHL).
ALPS, which should be taken into account in the evaluation of unusual cases (53,87,88,89). A subset of SHML shows features of IgG4-related disease, and an overlap between certain aspects of the two diseases has been suggested (90).
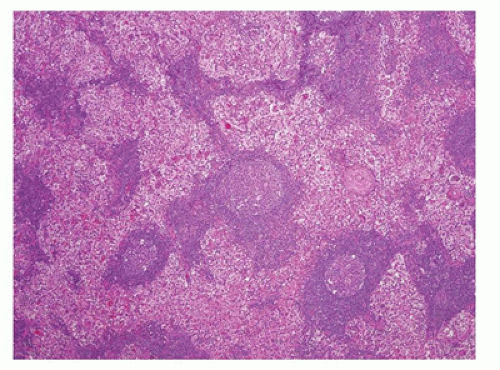 FIGURE 23-14 • As a result of massive sinusoidal expansion by histiocytes, germinal centers are compressed in Rosai-Dorfman disease (SHML). (Hematoxylin and eosin stain 4× magnification.) |
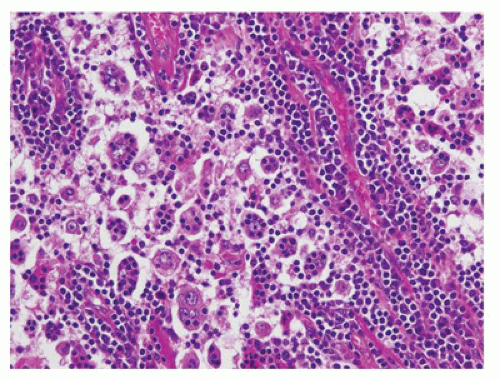 FIGURE 23-15 • The lesional cell in Rosai-Dorfman disease (SHML) has a small, cytologically bland nucleus and abundant eosinophilic cytoplasm containing one or more lymphocytes (emperipolesis). Although obvious in this case, emperipolesis may be difficult to detect on routine sections. (Hematoxylin and eosin stain 20× magnification.) |
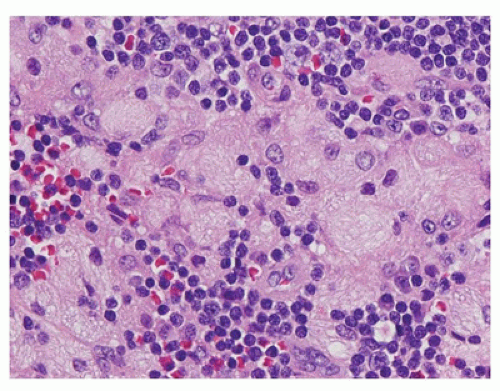 FIGURE 23-16 • Histiocytic proliferations caused by congenital storage disorders have a sinusoidal, often paracortical distribution in lymph nodes. The histiocytes seen in storage disorders often have coarsely vacuolated, (“bubbly”) or fibrillar (“crumpled tissue paper”) cytoplasm, as seen in this case of Gaucher disease. (Hematoxylin and eosin stain 40× magnification.) |
may have a pink mottled appearance because of infiltrating histiocytes and Langerhans cells (S100+, CD1a+, CD207+), some of which may contain coarsely granular brown-black melanin pigment (melanophages) that is positive on Fontana-Masson staining. Occasional hemosiderin pigment is also seen. FNA of lymph nodes with dermatopathia shows large clusters of histiocytes, histiocytes with melanin pigment, few tingible body macrophages, and histiocytes with elongated or grooved nuclei (94).
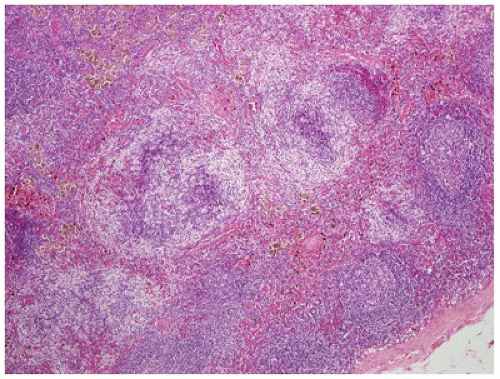 FIGURE 23-17 • The paracortical regions in dermatopathic lymphadenitis are expanded and show a pale pink swirled appearance due to the collections of abundant histocytes and Langerhans cells admixed with small lymphocytes. Some histiocytes contain brown melanin pigment. (Hematoxylin and eosin stain 4× magnification.) |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


