Chapter 8 THE LIVER AND BILIARY SYSTEM
Introduction
 Liver diseases account for more than 40,000 deaths annually in the United States alone.
Liver diseases account for more than 40,000 deaths annually in the United States alone.



 Adverse drug reactions involve the liver in a significant number of cases.
Adverse drug reactions involve the liver in a significant number of cases.

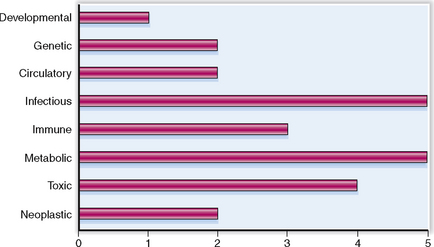
Liver diseases may have many causes. The relative clinical significance of various liver diseases is presented graphically in Figure 8-1.
Anatomy and Physiology
Bilirubin Yellow pigment derived from the heme portion of hemoglobin. Oxidation of heme gives rise to biliverdin, which is converted to bilirubin. Biochemically it is composed of four pyrrole rings (“open tetrapyrrole”). This unconjugated bilirubin is water-insoluble. It is bound to albumin and thereby transported to the liver. In the liver it is conjugated by UDP-glucuronyltransferase and thus made water-soluble. Through bile it is excreted into the intestine, where it is transformed by bacteria into urobilinogen and partially recirculated through the blood back to the liver. Conjugated bilirubin is referred to as direct and unconjugated as indirect. Bilirubin accounts for the yellow discoloration of the skin and mucosae in jaundice.
Common bile duct Main extrahepatic bile duct. It is a continuation of the common hepatic duct from the point where that duct is joined by the cystic duct to the orifice of the common bile duct in the duodenum at the papilla of Vater. It serves as a conduit for bile.
Gallbladder Hollow organ attached by the cystic duct to the main extrahepatic biliary ducts. It serves as a storage reservoir for bile.
Glisson’s capsule Peritoneum with underlying fibrous tissue enveloping the liver. Fibrous strands extend from the capsule into the liver parenchyma along the biliary ducts and blood vessels. Glisson’s capsule contains sensory nerves.
Hepatic artery Artery originating from the celiac trunk and providing most of the arterial blood to the liver.
Hepatic lobule Term for the hypothetical anatomic unit of the liver composed of a centrally located central vein surrounded by hepatocytes. Portal tracts containing the smallest branches of hepatic artery, portal vein, and bile ducts are located on the periphery of the lobule.
Hepatic vein Vein that drains venous blood from the liver into the inferior vena cava.
Hepatocyte Also known as the liver cell, it represents the principal metabolic cell in the liver.
Kupffer cell Fixed macrophage of the liver found in the hepatic sinusoids.
Papilla of Vater Small duodenal mucosal elevation at the site of entry of the common bile duct and pancreatic duct into the intestine.
Porta hepatis Also known as the hepatic hilum, it is the part of the visceral (inferior) surface of the liver through which the major blood vessels and extrahepatic bile duct enter the liver parenchyma.
Portal tract Triangular fibrous area at the periphery of the hepatic lobule that contains the terminal branches of the portal vein and hepatic artery, as well as the small biliary ducts. The limiting plate represents the sharp line separating the portal tract from the hepatocytes in the lobule (acinus).
Portal vein Vein formed through the confluence of the splenic and pancreatic superior and inferior mesenteric veins. It drains the venous splanchnic blood into the liver.
Sinusoids Small hepatic blood vessels, corresponding to capillaries. They differ from capillaries in that they are lined by a discontinuous (“fenestrated”) endothelial layer.
Space of Disse Narrow space between the apical surface of hepatocytes and the endothelial cells lining the sinusoids.
Sphincter of Oddi Smooth muscle bands encircling the terminal part of the common bile duct, common pancreatic duct, and ampulla of Vater. Cholecystokinin and neural stimuli cause relaxation or constriction of the sphincter, thus regulating the flow of bile into the intestine.
Stellate cells of Ito Stromal cells scattered at random in the spaces of Disse. They accumulate fat and vitamin A and may transform into collagen-producing myofibroblasts in cirrhosis.
Terminal hepatic venule Also know as the central vein of the lobule, it represents the beginning of the hepatic venous system inside the lobule (acinus). It is located at the center of the lobule. It receives the blood from hepatic sinusoids and drains that blood toward the hepatic vein and the inferior vena cava.
Pathophysiology and Laboratory Medicine
Ascites Accumulation of serous fluid in the abdominal cavity. Usually it is a sign of cirrhosis, but it may be also be a consequence of generalized hypoalbuminemia or chronic heart failure.
Cholecystitis Inflammation of the gallbladder. It is often associated with gallstones or bacterial infection.
Cholelithiasis Formation of gallstones in the hepatobiliary system, most often in the gallbladder.
Cholestasis Stagnation of bile inside the biliary tree usually due to biliary obstruction. Canalicular cholestasis may be a consequence of liver cell injury and is one of the well-known hepatic signs of injury. Cholestasis is associated with conjugated hyperbilirubinemia and bilirubinuria and may be accompanied by clay-colored (acholic) stools.
Fatty liver Also known as steatosis, it is a consequence of fat accumulation inside the hepatocytes. Fat begins to accumulate in the form of small droplets (microvesicular steatosis), which become confluent and eventually fill the entire liver cell with triglycerides (macrovesicular steatosis). The most common causes of fatty liver are obesity, diabetes, and alcohol abuse. Viral hepatitis C and some drugs (e.g., tetracycline) may also cause hepatic steatosis. Steatosis is a reversible cell change, but in some cases it may be accompanied by inflammation (steatohepatitis).
Gynecomastia Enlargement of male breast. In patients with cirrhosis it is usually caused by hyperestrinemia, resulting from incomplete hepatic inactivation of endogenous estrogens.
Hepatic encephalopathy Brain disturbance caused by liver disease. Symptoms include asterixis (coarse tremor and flapping of hands), loss of coordination, and progressive coma. Coma is graded on a scale from I to IV and may be lethal.
Hepatomegaly Enlargement of the liver. It may a consequence of various metabolic disturbances leading to accumulation of fat (e.g., obesity or diabetes), hemosiderin (e.g., hemochromatosis), or congestion of the liver in heart failure. Tumors may also cause hepatomegaly.
Hepatorenal syndrome Oliguric renal failure seen in patients with end-stage liver disease (cirrhosis) accompanied by ascites.
Jaundice (Latin, icterus) Yellow discoloration of the skin and mucosae that develops due to hyperbilirubinemia. It reflects the deposition of bilirubin in tissues. Jaundice becomes evident when the blood concentration of bilirubin exceeds 2 mg/dL.
Kernicterus Cerebral dysfunction caused by the deposition of bilirubin in basal ganglia. Typically it is caused by hemolytic anemia in infants due to maternofetal blood group incompatibility or severe prolonged hyperbilirubinemia, as in Crigler-Najjar syndrome type I.
Liver function tests (LFTs) Laboratory tests performed on blood to estimate the extent of liver cell injury and synthetic and excretory functions of the liver. They include measurements of serum transaminases (AST and ALT), alkaline phosphatase, bilirubin, albumin, and coagulation parameters (most often prothrombin time—PT).
Portal hypertension Elevation of blood pressure over 12 mm Hg in the portal system. The main consequences of portal hypertension are ascites, esophageal varices, and splenomegaly.
Splenomegaly Enlargement of the spleen. In cirrhotic patients it is usually related to chronic passive congestion that develops due to portal hypertension. It may be associated with signs of hypersplenism, including anemia and thrombocytopenia.
Vascular spider Also known as spider telangiectasia it represents a dilatation of small dermal blood vessels. It has a red central dotlike bulge with small branches radiating from it in all directions. Because of its resemblance to spiders it is also called spider nevus. It usually develops on the skin of the upper chest and, like palmar erythema, is a complication of hyperestrinism often found in cirrhosis.
Xanthoma Yellow skin papule, plaque, or nodule caused by hyperlipidemia. It is a feature of primary biliary cirrhosis and is associated with hypercholesterolemia. It may be found in primary disorders of lipid metabolism. If found on the palpebrae it is called xanthelasma.
Liver Diseases
Abscess of the liver Localized purulent inflammation of the liver caused by bacteria. The infection may reach the liver through the bile ducts (cholangitic abscess) or the branches of the portal vein (pylephlebitic abscess).
Acute hepatitis Inflammation of the liver, characterized by sudden-onset jaundice or other symptoms of relatively short duration.
Alcoholic liver disease Spectrum of diseases related to alcohol abuse, including fatty change, alcoholic steatohepatitis, and alcoholic cirrhosis. Fatty liver is almost always found in patients after excessive drinking of alcohol. Alcoholic hepatitis and cirrhosis develop in a minority of chronic alcohol abusers.
α1-Antitrypsin deficiency Genetic disease characterized by liver and pulmonary diseases. The affected liver may show signs of chronic hepatitis or cirrhosis. Symptoms may appear in any age group. This genetic defect is the most common cause of neonatal hepatitis.
Autoimmune hepatitis Autoimmune disease predominantly affecting young women. Associated with antismooth muscle antibodies (ASM), antinuclear antibodies (ANAs), and other autoimmune diseases. It responds to corticosteroid treatment but may also persist and progress to cirrhosis.
Budd-Chiari syndrome Syndrome caused by thrombosis of the hepatic vein and massive enlargement of the liver due to congestion. Most often it is a complication of such hematologic diseases as polycythemia, leukemia, and thrombophilia.
Cholangitis Inflammation of intrahepatic or extrahepatic bile ducts. It may be suppurative owing to bacterial infection, or nonsuppurative as in various autoimmune diseases (e.g., primary biliary cirrhosis).
Chronic hepatitis Chronic inflammation of the liver, most often caused by viral hepatitis C. It may persist and be relatively asymptomatic or it may progress to cirrhosis. Similar changes can be seen in various immune diseases of the liver and drug-related liver diseases.
Cirrhosis Chronic liver disease causing liver failure. The liver is of abnormal size and shape and subdivided into small nodules by abundant collagenous connective tissue. Cirrhosis may have many causes, the most important of which are chronic alcohol abuse and chronic viral hepatitis C infection. The cause of cirrhosis cannot be established in 15% to 20% of patients, and in such cases it is called cryptogenic.
Crigler-Najjar syndrome Hereditary unconjugated hyperbilirubinemia caused by absolute or relative deficiency of glucuronosyltransferase in liver cells.
Dubin-Johnson syndrome Hereditary conjugated hyperbilirubinemia, presenting as mild jaundice. It is caused by a blockage in bilirubin excretion from hepatocytes owing to the defect in the function of the ATP-binding cassette (ABC) of the canalicular multispecific organic anion transporter protein.
Gilbert syndrome Genetic disorder characterized by recurrent bouts of jaundice caused by unconjugated hyperbilirubinemia. It is related to mutation of the gene encoding uridine glucuronosyltransferase.
Hereditary hemochromatosis Genetic disease related to mutations of the HFE gene encoding the regulator of iron absorption in the small intestine. It is characterized by excessive absorption of iron from food. Excess iron is stored in the body, damaging multiple tissues. The most common complications of iron storage are cirrhosis, diabetes, hyperpigmentation of the skin, arthropathy, and cardiomyopathy.
Liver tumors Tumors of the liver can be classified as benign or malignant. The most common benign tumor is hemangioma. Other benign tumors are hepatocellular adenoma and focal nodular hyperplasia. Malignant tumors may be primary or metastatic. Primary tumors of the liver are hepatocellular carcinoma, cholangiocarcinoma, and angiosarcoma. Metastases to the liver can occur from any other primary site. Metastases are the most common malignant tumors of the liver.
Nonalcoholic steatohepatitis Chronic liver disease of unknown origin characterized by fatty change of hepatocytes, intralobular fibrosis, and focal infiltrates of inflammatory cells. It may cause portal hypertension and progress to cirrhosis.
Primary biliary cirrhosis Autoimmune liver disease primarily affecting women. It is a nonsuppurative cholangitis leading to destruction of bile ducts and progressive jaundice and cirrhosis. Other symptoms include pruritus, xanthelasma, and steatorrhea. Antibodies to mitochondria are a clue to the diagnosis.
Primary sclerosing cholangitis Disease of unknown origin but considered to be immune in nature. In 60% to 70% of cases it is associated with ulcerative colitis. The diagnosis is made by cholangiography, which shows typical “sausage-like” narrowing and dilatation of intrahepatic and extrahepatic bile ducts. Liver biopsy shows concentric fibrosis around large- and medium-sized bile ducts. The disease has a tendency to progress to cirrhosis.
Wilson’s disease Rare autosomal recessive genetic disorder characterized by accumulation of copper in the liver and other tissues. Cirrhosis develops together with degeneration of basal ganglia of the brain, resulting in Parkinsonism. A brownish green ring (Keyser-Fleischer ring) can be seen by slit lamp examination on the limbus of the cornea.
Normal Structure and Function
ANATOMY
The liver is located in the right upper abdominal quadrant underneath the diaphragm and mostly behind the rib cage.
The liver is located in the right upper quadrant of the abdomen behind the lower part of the rib cage (Fig. 8-2). Cranially its superior surface is in contact with the diaphragm. From the clinical point of view these anatomic facts are important for the following reasons:
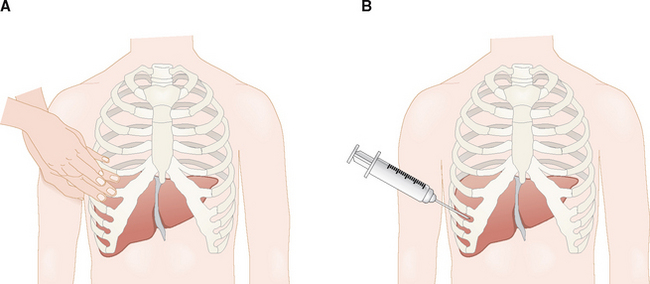
 The liver can be localized by percussion through the chest wall. In contrast to the resonant sound of the percussed lung, over the liver the percussion produces a dull sound. The distance between the upper and lower border of this dullness is used to estimate the size of the liver.
The liver can be localized by percussion through the chest wall. In contrast to the resonant sound of the percussed lung, over the liver the percussion produces a dull sound. The distance between the upper and lower border of this dullness is used to estimate the size of the liver.

 Percutaneous liver biopsy is performed through the intercostal space.
Percutaneous liver biopsy is performed through the intercostal space.
The size and the shape of the liver are relatively constant but may change under pathologic conditions.
The size of the liver depends on body size. On average, top to bottom it has a span of less than 13 cm when measured in the midcostal line. The size of the liver can be estimated by percussion, or by combining percussion and auscultation. Unfortunately these techniques lack precision and reproducibility. More accurate measurements can be made by ultrasonography or radiologic imaging. Ultrasonography may be also used to determine whether the liver has a normal shape and smooth surface, is irregularly shaped, or contains irregular masses. These abnormalities are visible in computed tomography (CT) scans as well.
Pathologically altered livers can be of normal size, enlarged, or reduced in size.
 Liver of normal size. In the course of most diseases, the size of the liver does not change significantly.
Liver of normal size. In the course of most diseases, the size of the liver does not change significantly.


The liver has relatively limited mobility and is linked through the bile ducts with the intestine.
The liver is enclosed by folds of the peritoneum, which together with the falciform ligament keep it fixated in the subdiaphragmatic position. The peritoneum covering the liver is known as Glisson’s capsule. On the visceral side of the liver the peritoneum covers the hilar structures (i.e., the gallbladder) and forms the lesser omentum surrounding the portal vein, hepatic artery, and the extrahepatic biliary ducts (Fig. 8-3).
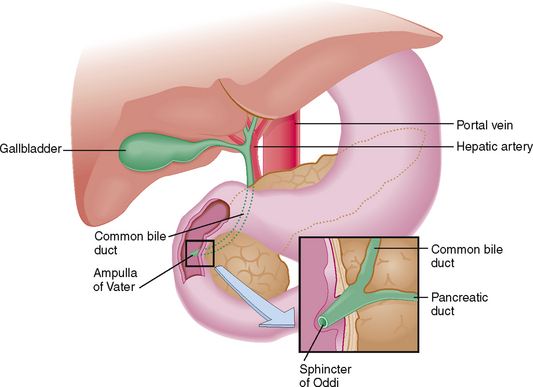
> Glisson’s capsule contains sensory nerves.
> Venous congestion of the liver causes expansion of the capsule and is accompanied by pain.
> Do not forget to anesthetize the capsule prior to liver biopsy.
The liver has a dual blood supply.
The liver has a dual blood supply. The hepatic artery, a branch of the hepaticoduodenal artery originating from the celiac axis, provides the arterial blood, which accounts for 25% to 30% of the total hepatic blood supply. The portal vein, a large valveless vein draining the venous blood from the intestines, stomach, pancreas, and spleen, brings in the remaining 70% to 75% of the blood. The venous blood leaves the liver through the hepatic veins, which drain into the inferior vena cava.
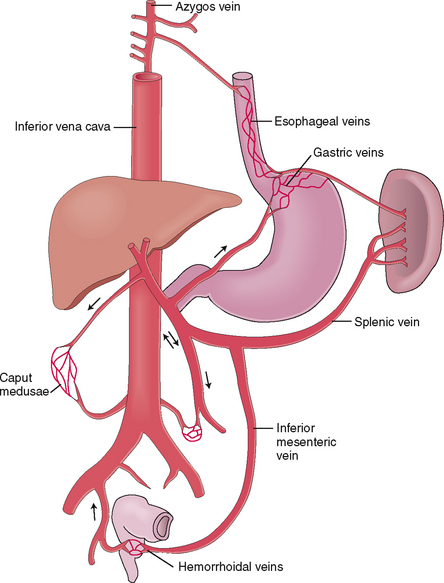
In most instances portal hypertension is a consequence of liver disease. If the blood cannot be drained from the portal system through the liver and the portal blood pressure exceeds 20 mm Hg, anastomoses, or collaterals, develop between the portal and systemic venous system. These anastomoses develop through the dilatation and reopening of small veins that normally connect the portal and systemic venous systems. Under normal circumstances these small veins contain very little blood, but in portal hypertension they transform into congested, tortuous, widely opened venous channels that can be visualized by angiography. These anastomotic collaterals most often develop in the area of the lower esophagus and gastric fundus, internal hemorrhoidal veins and retroperitoneum, and the periumbilical veins (Fig. 8-4). The dilatation of periumbilical veins is traditionally called caput Medusae, in reference to the Gorgon from Greek mythology whose hair was made of snakes.
HISTOLOGY
Hepatocytes perform most complex liver functions.
Hepatocytes, or liver cells, are polarized cells interconnected along their apical sides with intercellular junctions. Tight junctions are important for the maintenance of liver cell polarity; they also ensure the structural stability of liver cell cords. In the midportion of the apical surface two adjacent liver cells form intercellular canaliculi, which are filled with bile secreted by the hepatocytes. The cell surface that does not have tight junctions is called the basolateral side. It delimits the space of Disse, which on the other side is separated from the lumen of the sinusoids by endothelial and Kupffer cells (Fig. 8-5).
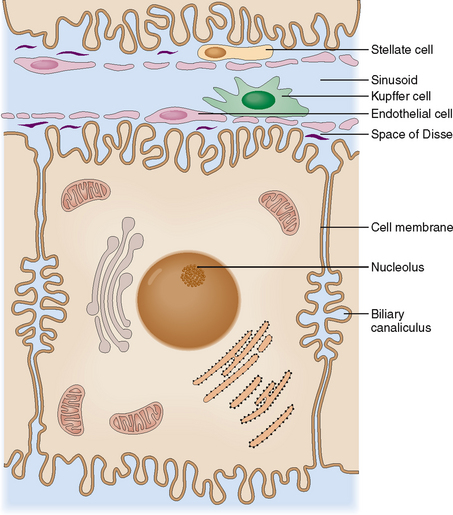




Sinusoids are essential for the normal exchange of metabolites between the blood and liver cells.
In addition to the endothelial cells, the sinusoids contain scattered Kupffer cells, which act as fixed macrophages. These cells have major scavenger functions and participate in the removal of particulate material, bacteria, and immune complexes from the circulation. Inside the spaces of Disse are scattered the stellate cells of Ito, which have a capacity to store lipids and, if properly stimulated, also synthesize collagenous extracellular matrix. These cells play a major role in the formation of fibrous tissue in liver cirrhosis.
The biliary ducts and the gallbladder are lined by cuboidal to cylindrical cells specializing in the excretion of bile.
The biliary excretory system begins with intercellular bile canaliculi on the apical (intercellular) surface of the hepatocytes (Fig. 8-6). From these canaliculi the bile flows into minor bile ducts in the portal tracts as well as medium-sized and larger (septal) bile ducts draining the bile toward the hepatic bile ducts in the porta hepatis. All bile ducts are lined by cuboidal to cylindrical cells lying in a polarized manner on a basement membrane. Similar polarized cells line the major extrahepatic bile ducts all the way to the papilla of Vater. These cells produce some components of the bile and are also important for the maintenance of bile outflow from the liver. The cells lining the gallbladder resemble those in the bile ducts. However, gallbladder cells are unique in that they can actively absorb sodium and chloride from the bile. The resorption of sodium chloride is followed by a passive outflow of water and concentration of bile.
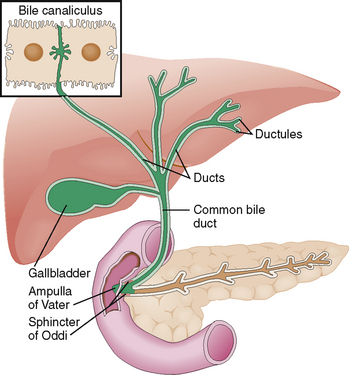
Connective tissue extends along the bile ducts and blood vessels all the way to the portal tracts in the center of hepatic acini.
Liver cells are arranged into functional units called lobules or acini.
All hepatocytes are interchangeable among themselves and thus all of them can perform all hepatic functions. Under normal circumstances the function of each hepatocyte nevertheless depends on it location in the hexagonal microscopic unit called the lobule or acinus (Fig. 8-7).
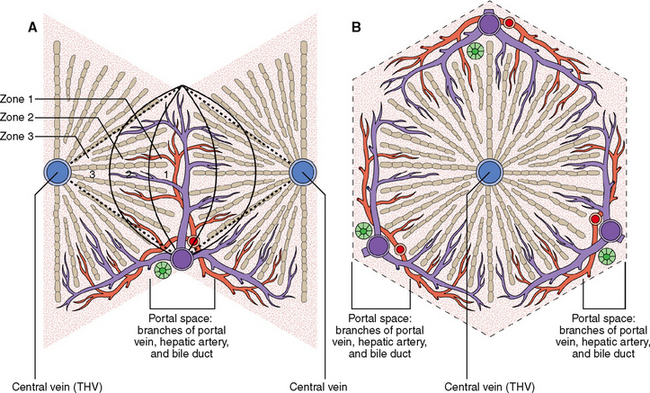
The hepatocytes located along these pressure gradients have different functions, probably determined by the oxygen supply (which is higher around the portal tracts than around the THV) and the concentration of nutrients and metabolites. The acinus can be thus divided into three functional zones: (1) periportal zone 1, (2) perivenular zone 3, and (3) zone 2 in between zones 1 and 3. Furthermore it was shown that the falling blood pressure from the center of the acinus toward its periphery is accompanied by a decreasing concentration of oxygen in hepatic blood and a decreased concentration of various metabolites and toxins. For example, ammonia is mostly extracted from portal blood as soon as it enters into the acinus (zone 1) so that very small amounts of this substance reach the perivenular periphery of the acinus. The hepatocytes of zone 1 are more active in oxidative phosphorylation and gluconeogenesis than those in zone 3. Perivenular hepatocytes, adapted to relative hypoxia, tend to engage more in anaerobic glycolysis and lipogenesis.




Pathophysiology
Since almost all functions of the liver cells are performed simultaneously and are thus interconnected, the answer might not be so simple. For didactic purposes we, nevertheless, discuss each of the major functions separately and outline the most important consequences of liver cell dysfunction, including the following processes:
 Intermediate metabolism of carbohydrates, lipids, and proteins
Intermediate metabolism of carbohydrates, lipids, and proteins

 Detoxification of endogenous and exogenous substances
Detoxification of endogenous and exogenous substances
 Bilirubin metabolism and excretion
Bilirubin metabolism and excretion

DISTURBANCES OF INTERMEDIATE METABOLISM
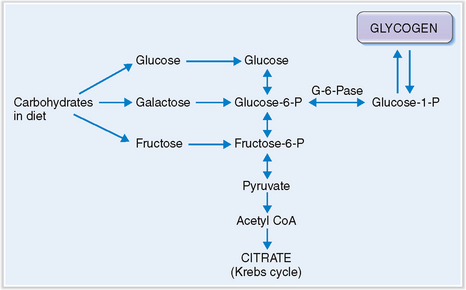
Glycogen accumulates in the liver of infants who lack glucose-6-phosphatase.
The glucose taken up from blood is stored in the liver in the form of glycogen. Glycogen stores (70–80 g) are sufficient to meet the body’s demand during 24 hours of fasting, after which gluconeogenesis from amino acids becomes the primary source of glucose. In children who have von Gierke’s disease, or glycogenosis type I, the hepatocytes lack glucose-6-phosphatase and therefore cannot form glucose to be exported into the blood (Fig. 8-8). Such children suffer from hypoglycemia. At the same time glucose-1-phosphate accumulates, promoting glycogen accumulation in liver cells and resulting in hepatomegaly. Compensatory metabolic changes lead to hyperlipidemia and lactic acidosis.
Lipids can accumulate in liver cells due to increased supply and influx, increased endogenous lipogenesis, lowered utilization, or decreased excretion.
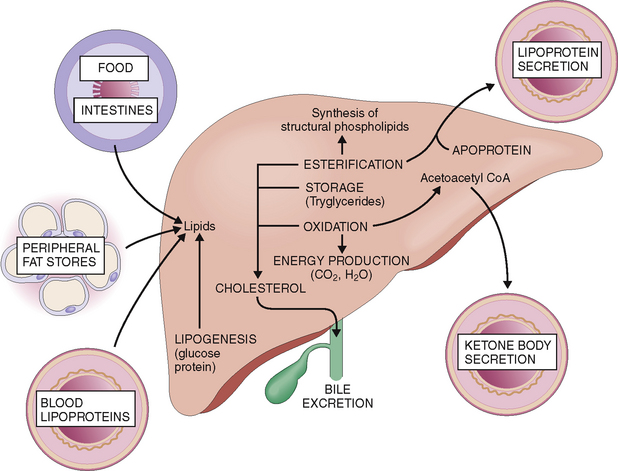
Lipids are transported by blood to the liver from food absorbed in the intestines or from fat stores and other tissues (Fig. 8-9). The fat absorbed from food in the intestines is packaged into chylomicrons, which enter the intestinal lymph and from there enter the blood. During the passage of such blood through the small blood vessels of the skeletal muscle and fat tissue, the endothelial lipoprotein lipase acts on the chylomicrons, resulting in the formation of glycerol, free fatty acids, and cholesterol-enriched chylomicron remnants. Most of the glycerol and fatty acids thus formed are absorbed by muscle cells and fat cells, whereas the chylomicron remnants reach the liver and are taken up through the low-density lipoprotein (LDL) and LDL-related receptors.
 Oxidized and used for production of energy.
Oxidized and used for production of energy.


 Used for synthesis of cholesterol, which may be further used for endogenous purposes, excreted in bile, metabolized to bile acids and excreted in bile, or packaged into very low density lipoproteins (VLDLs) and secreted into the blood.
Used for synthesis of cholesterol, which may be further used for endogenous purposes, excreted in bile, metabolized to bile acids and excreted in bile, or packaged into very low density lipoproteins (VLDLs) and secreted into the blood.

Fatty liver (steatosis) can be induced by increasing the supply of lipids by overeating. Obesity, diabetes, and alcoholism are also associated with overabundance of lipid influx into the liver. Alcohol mobilizes free fatty acids from peripheral fat tissue stores, but it also promotes the esterification of intrahepatic fatty acids into triglycerides, and it inhibits the synthesis of the apoproteins essential for synthesis and export of VLDLs. Starvation and protein-deficient malnutrition may cause fatty liver due to inadequate synthesis and export of lipoproteins. The most important causes of fatty liver are listed inTable 8-1.
Table 8-1 Common Causes of Fatty Liver
Protein synthesis is a major function of liver cells.
Liver cells synthesize proteins for endogenous purposes but also for export. Proteins for exogenous purposes are synthesized in the cisterns of the rough endoplasmic reticulum, glycosylated or folded, and actively secreted into the blood. Most of the plasma proteins are synthesized in the liver. Liver disease results in marked reduction of plasma protein synthesis, which is usually associated with significant pathophysiologic changes. For example, hypoalbuminemia occurs in chronic liver disease, resulting in diminished oncotic pressure of the plasma and predisposing to edema formation.
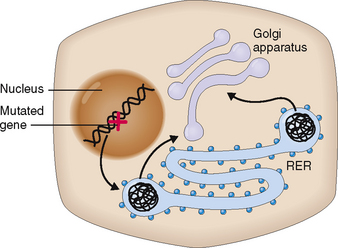
Abnormal synthesis of some proteins may also cause structural changes in hepatocytes, which are visible by light and electron microscopy. The best example is α1-antitrypsin (AAT) deficiency, an autosomal recessive disorder characterized by the inability of liver cells to excrete AAT. The defect lies in the abnormal folding of the AAT in the cisterns of the rough endoplasmic reticulum of hepatocytes, which cannot then complete the synthesis of the protein and retains the abnormal intermediate product inside the cytoplasm in the form of round globules (Fig. 8-10). Deficiency of AAT predisposes the affected person to cirrhosis but also to pulmonary emphysema.
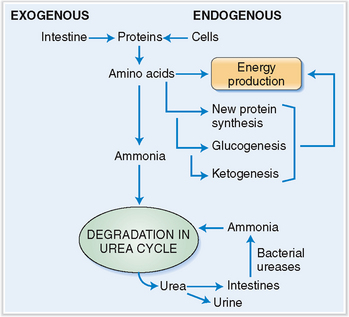
Amino acids are metabolized to urea and ammonia, which may be toxic.
Amino acids absorbed from the intestines in surplus to the needs of the body as well those that are released after normal cell turnover are used for the production of energy, for the synthesis of new proteins, or for ketogenesis or glucogenesis, or are metabolized further into urea (Fig. 8-11). Urea is excreted in urine and feces. Urea that is excreted in kidneys leaves the body, but the urea excreted into the intestine is again cleaved by urease-containing bacteria, and the newly formed ammonia is absorbed into the portal circulation and sent back to the liver. Approximately 10 to 20 g of nitrogen are produced every 24 hours in an average adult and excreted.
DISTURBANCES OF HEPATIC SYNTHETIC FUNCTIONS
Chronic liver disease leads to decreased synthesis of serum albumin and hypoalbuminemia.
Hypoalbuminemia may be aggravated in patients with portal hypertension by excessive leakage of albumin from the blood into the ascites fluid or the lymph away from its normal flow through the liver. Hypoalbuminemia may also be partly related to poor nutrition, which is especially common in cirrhotic patients who are chronic alcoholics. Finally, bear in mind that albumin is a “negative acute-phase reactant”; that is, the liver reduces the synthesis of albumin in response to many acute and chronic diseases. Since patients with advanced cirrhosis usually feel sick, this is yet another reason why they might have hypoalbuminemia.
DISTURBANCES OF DETOXIFICATION
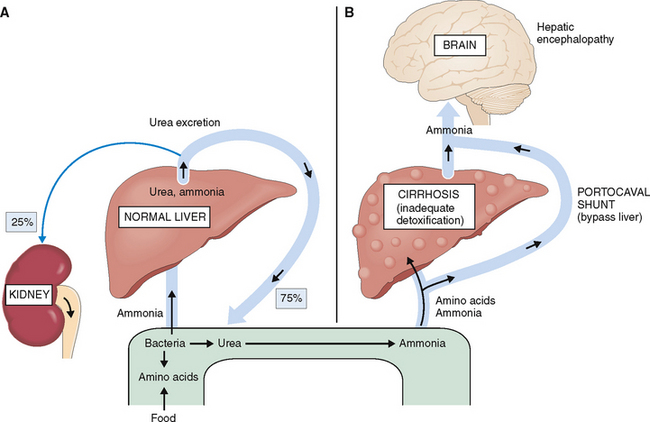
The liver degrades and neutralizes ammonia, thus disabling its toxicity.
Cirrhosis and massive liver necrosis may reduce the capacity of the liver to remove ammonia formed from the degraded amino acids. In cirrhotic patients with portal hypertension the blood bypasses the liver through the portal-systemic anastomoses, further contributing to body’s inability to remove ammonia. If the portal hypertension-related esophageal varices rupture and the patient has a massive bleed, the swallowed blood becomes yet another source of ammonia. Blood is a protein-rich fluid, and when it arrives into the intestines, the proteins are degraded into amino acids and further into ammonia. Hyperammonemia resulting from any of these complications of cirrhosis has a potentially toxic effect on the brain and is considered to play an important pathogenetic role in hepatic encephalopathy (Fig. 8-12).
DISTURBANCES OF BILIRUBIN METABOLISM AND EXCRETION
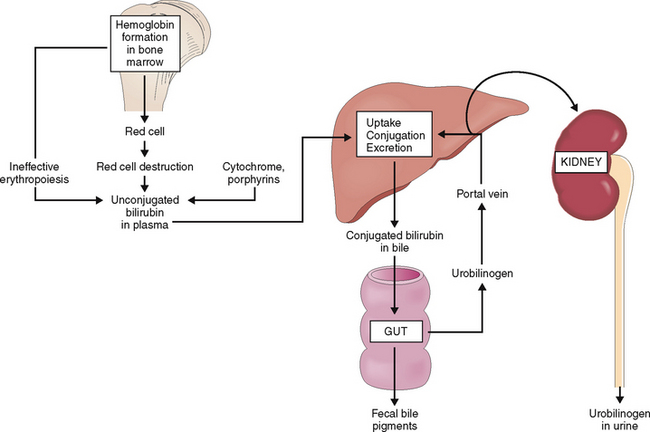
Bilirubin derived from the heme component of the hemoglobin is bound to albumin and carried to the liver.
Approximately 4 mg/kg of bilirubin is formed daily, mostly from the heme component of hemoglobin released from effete red blood cells (Fig. 8-13). A smaller part of the newly formed bilirubin (15% to 20%) is derived from ineffective hematopoiesis and heme-containing enzymes such as P450 oxidoreductases or other porphyrins.
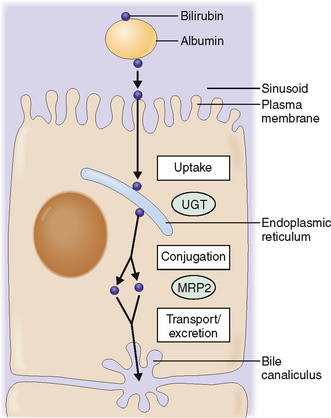
Bilirubin taken up by hepatocytes is conjugated to glucuronide.
The bilirubin–albumin complex binds to the basolateral side of hepatocytes. It dissociates from albumin and is actively transported across the plasma membrane into the cytoplasm of liver cells. At least three distinct mechanisms participate in this process. In the cytoplasm bilirubin is transferred into the endoplasmic reticulum, most likely by direct membrane-to-membrane transfer. In the endoplasmic reticulum bilirubin is then conjugated through the action of uridine disphosphate (UDP) and uridine glucuronyltransferase (UGT) into monoglucuronides and diglucuronides and readied for excretion (Fig. 8-14).
 Gilbert syndrome is an common periodic unconjugated hyperbilirubinemia that is related to mutations in the promoter region of the UGT1A1 gene or a missense mutation of the UGT1A1 gene itself.
Gilbert syndrome is an common periodic unconjugated hyperbilirubinemia that is related to mutations in the promoter region of the UGT1A1 gene or a missense mutation of the UGT1A1 gene itself.
 Crigler-Najjar syndrome is an unconjugated hyperbilirubinemia that occurs in two forms. Type I of the syndrome is characterized by severe congenital hyperbilirubinemia that has a high mortality rate in infancy. It is also related to the mutation of the UGT1A1 gene, resulting in the complete lack of the enzyme in liver cells. In Crigler-Najjar syndrome type II, the activity of UGT1A1 is markedly reduced, but it still can form monoglucuronides.
Crigler-Najjar syndrome is an unconjugated hyperbilirubinemia that occurs in two forms. Type I of the syndrome is characterized by severe congenital hyperbilirubinemia that has a high mortality rate in infancy. It is also related to the mutation of the UGT1A1 gene, resulting in the complete lack of the enzyme in liver cells. In Crigler-Najjar syndrome type II, the activity of UGT1A1 is markedly reduced, but it still can form monoglucuronides.
Bilirubin reaches the intestine through the bile ducts.
Bilirubin in bile has an average concentration of 0.2%, yet it gives the bile its typical yellow-brown color. Bile flows from the intercellular canaliculi into the bile ducts in the portal tracts and from there through larger bile ducts to the hilum of the liver and into the common bile duct. Obstruction of bile ducts causes regurgitation of bilirubin into the blood and conjugated hyperbilirubinemia. Clinically it manifests as obstructive jaundice. The most important causes of obstructive jaundice are illustrated in Figure 8-15.
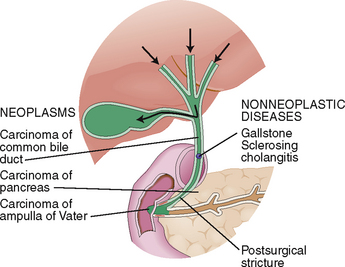
DISTURBANCES OF BILE CIRCULATION
The liver cells produce bile and excrete it into the bile ductules.
Bile is a complex bicarbonate-rich fluid produced by the liver cells and excreted through the biliary ducts into the intestines. Approximately 450 mL of canalicular bile is produced daily, which is supplemented with about 150 mL of ductular secretion to account for a total of 600 mL.
The bile is a complex fluid that consists principally of water (97%) and the following solutes:






Primary bile acids are produced in the hepatocytes from cholesterol and conjugated to glycine or taurine (Fig. 8-16). Once excreted from the liver cells, bile acids combine with cholesterol and phospholipids to form micelles, which are important for the emulsification and subsequent absorption of fat in the small intestine. Bile ductular cells secrete mucus, composed of proteins and carbohydrates, as well as water and minerals.
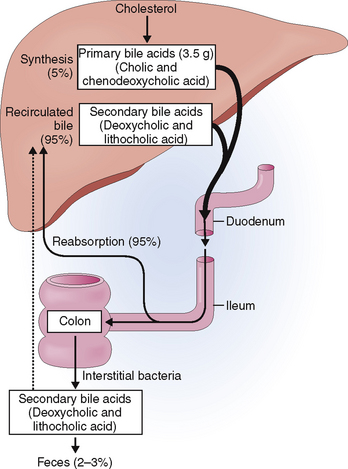
A large portion of this hepatic bile is extruded into the duodenum during the meals (Fig. 8-17). Between meals the sphincter of Oddi contracts and redirects the bile flow into the gallbladder, where it is concentrated by a removal of water and stored until needed.
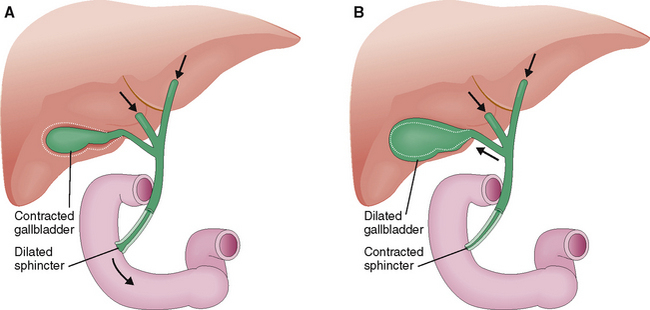
Changes in the composition of bile may predispose to formation of gallstones.
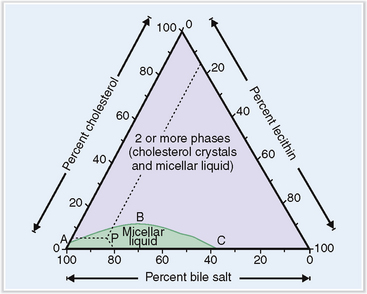
The solutes found in bile remain in a soluble form as long as the relative concentration of cholesterol, bile salt, and lecithin remains in the normal range (Fig. 8-18). Changes in the composition of bile and the presence of substances that promote “nucleation of bile” (e.g., bacteria) may lead to the formation of gallstones.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


