From 14 to 22 weeks, nephron arcades are formed, with the innermost nephron formed first (juxtamedullary nephron) (eFigure 17-1). From 22 to 36 weeks, no branching of the ureteric bud occurs. The ampulla extends to the subcapsular cortex to induce four to seven nephrons (eFigure 17-2). Thus, the nephrons formed last are subcapsular (the nephrogenic zone seen in sections of fetal kidneys) (Figure 17-1). From 36 weeks of gestation through birth and up to maturity, the nephrons grow, but no new nephrons are formed. In extremely premature infants, nephrogenesis continues after birth until the kidney reaches maturity. However, in this setting, renal maturation appears to be accelerated and associated with an increased number of morphologically abnormal and large glomeruli, suggestive of hyperfiltration and predicting fewer functioning nephrons in postnatal and later life (5).
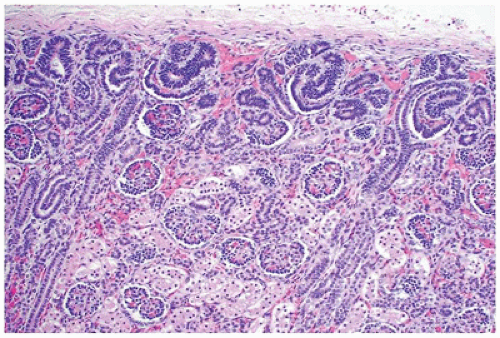 FIGURE 17-1 • Early third trimester kidney with subcapsular nephrogenic zone. (Hematoxylin-eosin stain, original magnification ×100.) |
the kidney has historically been based on morphologic criteria established decades ago; more recent systems attempt to incorporate new information about nephrogenesis and the pathogenesis of urinary tract malformations (10,11). It is well established that many cases of CAKUT have a genetic basis and many are associated with syndromes; nonsyndromic forms may also be causally linked to several developmental genes. Thus, neither approach is perfect as a given gene can result in a spectrum of anomalies, and the same anomaly can be caused by multiple genes. Nonetheless, it is worthwhile to consider the molecular mechanism of kidney and urinary tract development to better understand the malformation. The diversity of signaling pathways in nephrogenesis likely explains the remarkable locus heterogeneity found in CAKUT. Currently, mutations can be identified in approximately 10% to 20% of children, with HNF1B and PAX2 mutations being the most common (12). Table 17-3 lists the common malformations of the kidney and includes cystic diseases because many of these are inheritable disorders or are secondary to malformations of the kidney parenchyma and lower urinary tract (13).
TABLE 17-1 RENAL MALFORMATIONS SEN IN PEDIATRIC AUTOPSIES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
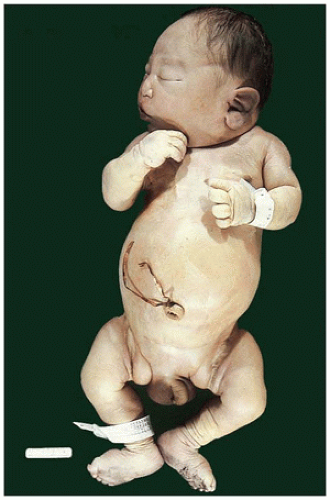 FIGURE 17-2 • Oligohydramnios (Potter sequence) is associated with low-set and deformed ears, beaked nose, receding chin, and lower limb positional deformity. |
anomalies of the urinary tract (14). While renal function is normal in the neonatal period in patients with renal ectopia without other associated malformations, vesicoureteral reflux and hydronephrosis have been reported in approximately 20% to 50%, particularly in crossed renal ectopia (16). Pseudocrossed renal ectopia occurs when an enlarging retroperitoneal mass displaces the kidney to the contralateral side of the abdomen.
TABLE 17-2 Renal Findings in Children with Oligohydramnios Sequence | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
TABLE 17-3 CLASSIFICATION OF MALFORMATIONS OF THE KIDNEY, INCLUDING CYSTIC DISEASES | |
|---|---|
|
more than half of patients with horseshoe kidney, secondary to ureteropelvic junction obstruction, reflux, or malrotation. Individuals with horseshoe kidney are at higher risk for the development of stones (19) and tumors (20). Rare cases of renal-adrenal fusion have been described, which may present as a renal mass in the upper pole (21).
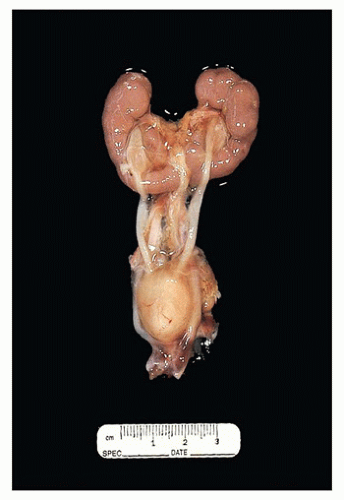 FIGURE 17-3 • Horseshoe kidney with fused lower poles. |
Long-term follow-up of patients with URA has shown that these patients are at higher risk for the development of proteinuria, hypertension, and renal insufficiency.
in the kidney, the principal clinical manifestations are caused by fetal and neonatal oliguria.
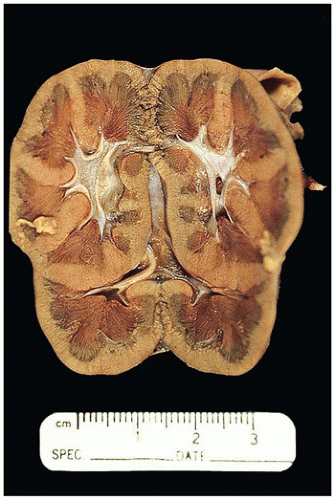 FIGURE 17-4 • Renal duplication (duplex kidney) with two separate pelves in the same kidney and more than the normal number of reniculi. |
seen (Figures 17-5 and 17-6). Neonatal hydronephrotic kidneys seen at autopsies usually have a histologically normal, although grossly compressed, cortex and medulla.
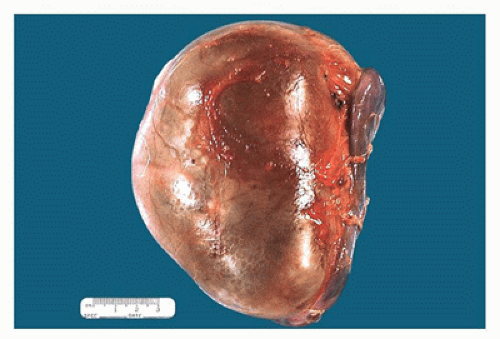 FIGURE 17-5 • Nephrectomy specimen from a 5-year-old who presented with a unilateral renal mass. The kidney appears to be one large cystic structure. |
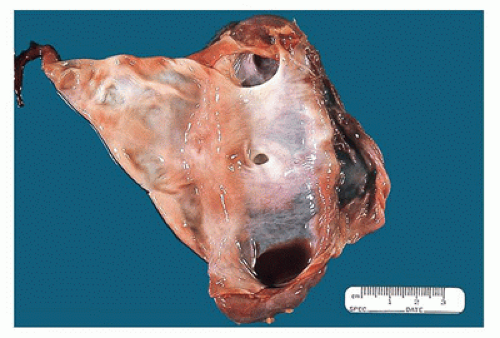 FIGURE 17-6 • Cut section of the kidney in Figure 17-5 shows a markedly dilated pelvis and calyces and very little residual renal parenchyma. |
appreciated through the capsule and on sectioning are seen to be irregularly distributed throughout the parenchyma, with no residual identifiable cortex or medulla (Figure 17-8).
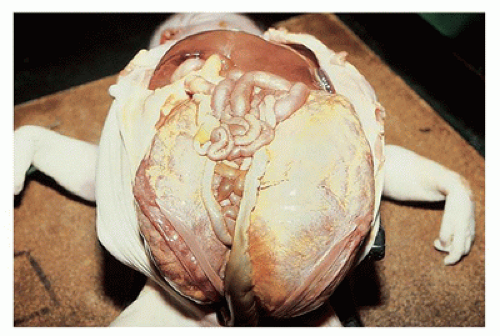 FIGURE 17-7 • Massively enlarged cystic dysplastic kidneys. |
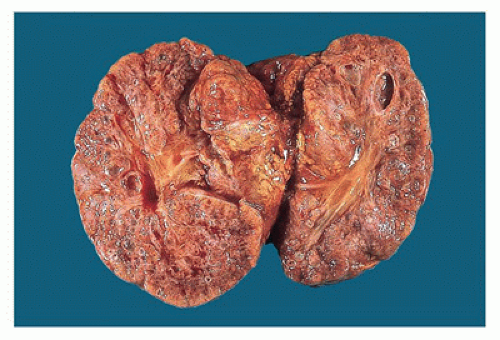 Figure 17-8 • Cut surface of cystic dysplastic kidney with multiple small, variably sized cysts involving both cortex and medulla. |
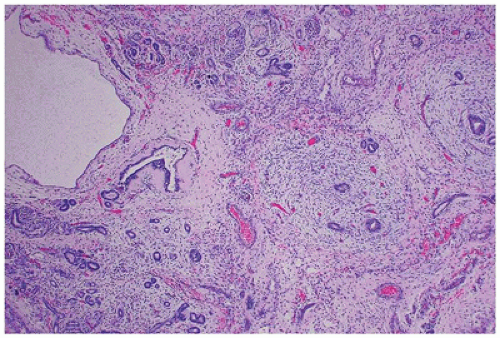 FIGURE 17-9 • Cystic dysplastic kidney with disorganized renal parenchyma in which immature tubules are surrounded by collarettes of condensed mesenchyme. (Hematoxylineosin stain, original magnification ×40.) |
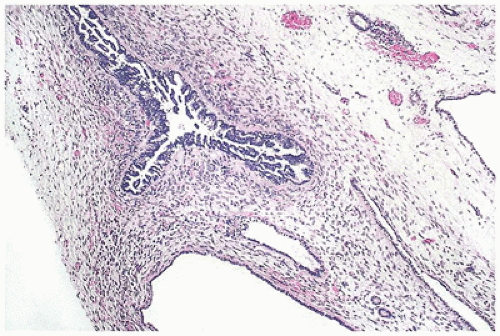 FIGURE 17-10 • Dysplastic tubules with an excessive amount of lining epithelium, which is thrown into papillary folds. (Hematoxylin-eosin stain, original magnification ×40.) |
majority of multicystic dysplastic kidneys (MCDK) can be an isolated finding and is uncommonly bilateral, it is often associated with other (contralateral) anomalies of the kidney and urinary tract. The vast majority of MCDK are associated with congenital urinary tract obstruction, which is often at the ureteropelvic junction but may occur at any level. Several animal models of renal dysplasia after gestational ureteral obstruction have been described (62).
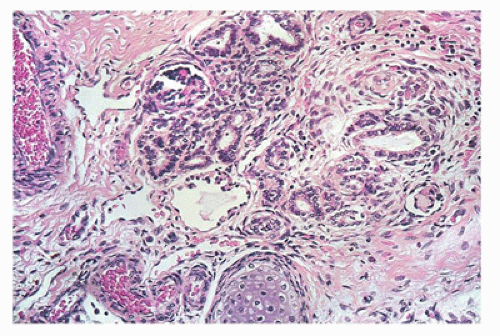 FIGURE 17-11 • Disorganized renal parenchyma and island of immature cartilage in cystic dysplastic kidney disease. (Hematoxylin-eosin stain, original magnification ×100.) |
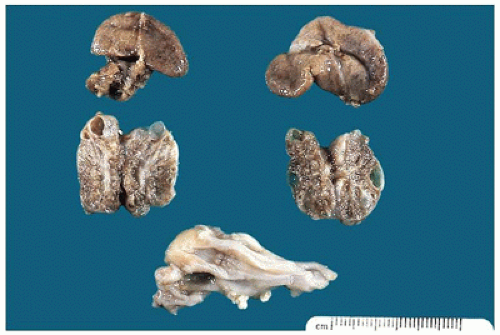 FIGURE 17-12 • Cystic dysplastic kidneys, shown bisected in the middle of the picture, are smaller than the adrenals above. |
TABLE 17-4 SYNDROMES AND DISEASES ASSOCIATED WITH RENAL DYSPLASIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
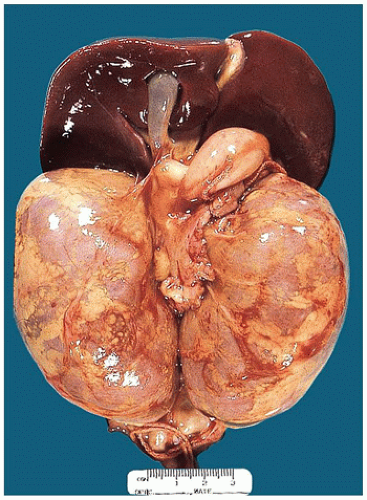 FIGURE 17-13 • Autosomal recessive polycystic kidney disease with massively enlarged symmetric reniform kidneys. |
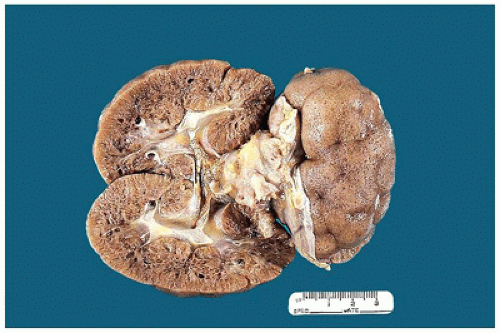 FIGURE 17-14 • Cysts of autosomal recessive polycystic kidney disease can be appreciated on the cortical surface. The cut section shows radially oriented cysts in the cortex and more rounded cysts in the medulla. |
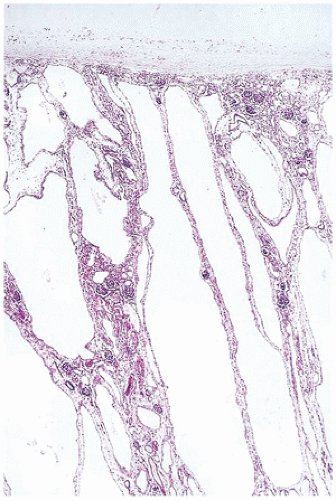 FIGURE 17-15 • Radially arranged cysts of autosomal recessive polycystic kidney disease. Normal glomeruli and tubules are seen between the cysts. (Hematoxylin-eosin stain, original magnification ×40.) |
protein. From these data, researchers have concluded that the milder mutation defines the phenotype. No significant clinical differences have been observed between patients with two missense mutations and those with a truncating mutation in trans to a missense mutation (67).
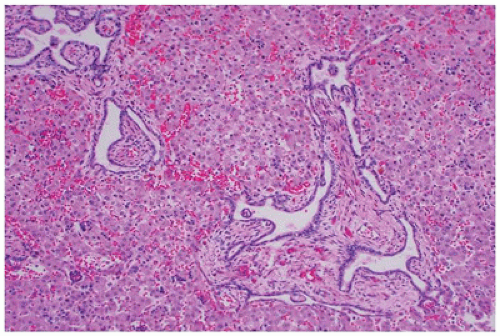 FIGURE 17-16 • Ductal plate malformation of the liver with expanded portal area, peripheral tortuous dilated bile ducts, and blood vessels in the middle. (Hematoxylin-eosin stain, original magnification ×40.) |
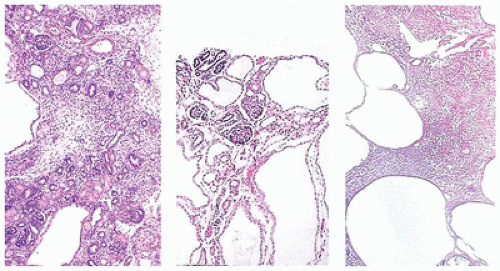 FIGURE 17-17 • Low-power photomicrographs illustrate the differences between cystic dysplastic kidney (left), autosomal recessive polycystic kidney disease (middle), and autosomal dominant polycystic kidney disease (right). (Hematoxylin-eosin stain, original magnification ×40.) |
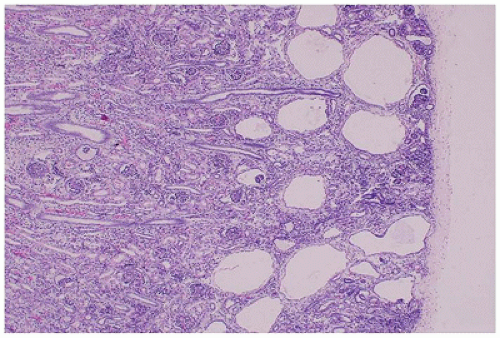 FIGURE 17-18 • GCKD with cystic dilatation of Bowman spaces; the medulla is uninvolved. (Hematoxylin-eosin stain, original magnification ×40.) |
is associated with mutations in TCF2, which encodes HNF1-β, and is considered the main cause of fetal hyperechoic kidneys (100) and, in some families, is associated with maturity-onset diabetes of the young, type V (MODY5) (101,102).Glomerular cysts in all types of GCKD are less than 1 cm in size and located in the cortex from the subcapsular zone to the inner cortex (eFigure 17-5), and are histologically similar to glomerular cysts seen in other disease conditions.
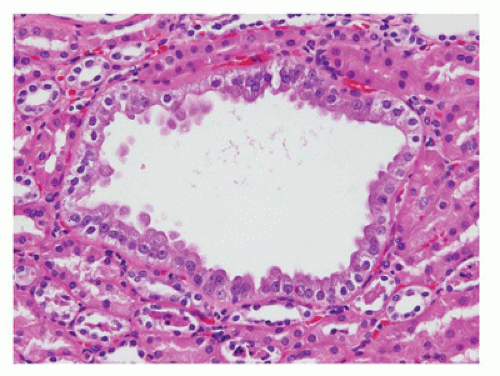 FIGURE 17-19 • Renal cysts of tuberous sclerosis lined by characteristic hyperplastic epithelium with abundant eosinophilic granular cytoplasm. (Hematoxylin-eosin stain, original magnification ×200.) (Courtesy of Dr. John Hicks, Houston, TX). |
organs (69). Multiple renal cysts and tumors develop in approximately two-thirds of patients with VHL disease (109). The most common imaging finding is multiple small unilateral or bilateral renal cysts, although renal function is usually retained (110). Less frequently, the cysts are numerous enough to simulate ADPKD. Renal cysts are lined by flattened clear epithelial cells; however, dysplastic microfoci of hyperplastic epithelium with clear cytoplasm and a “hobnail” appearance, often associated with complex cysts, are associated with a markedly increased risk for the development of renal cell carcinoma (RCC) (111). Multifocal cystic adenocarcinomas develop in 45% to 50% of patients beyond the third decade of life and in about 70% by the fifth decade (69,109). Most RCCs are of the clear cell type although clear cell papillary RCCs have been reported in patients with VHL disease (112).
Definitive identification of glomeruli may rarely require rapid frozen section, or the pathologist may be asked to perform a rapid frozen section to determine if crescents are present. In either case, the tissue submitted for frozen section can also be utilized for immunofluorescent (IF) studies. Whenever possible, tissue should be sampled for light, IF, and electron microscopy (EM), even if all those studies are not initially requested, and with the smaller-gauge biopsy needles now used by pediatric nephrologists, two or three cores are usually required. The specimen submitted for light microscopy (LM) should contain as much cortex as possible along with the corticomedullary junction, whereas only cortical tissue is ordinarily required in the specimens submitted for IF and EM (124).
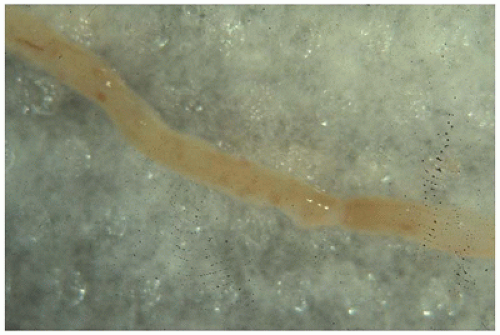 FIGURE 17-20 • Needle biopsy specimen of kidney viewed through a dissecting microscope. Glomeruli appear as red dots in the central region and vasa recta in the outer medulla as linear striations at either end. (Original magnification, 5×.) |
TABLE 17-5 TERMS USED IN DESCRIBING GLOMERULAR LESIONS | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
within mesangia (Figure 17-22B), or both (Figure 17-22C)—indicates immune complex (IC) deposition, and the site and the composition of the immunoreactant(s) depend on the disease. Crescents often stain brightly for fibrinogen. Linear staining along the capillary wall may indicate antiglomerular basement membrane disease (usually only IgG). Dense deposit disease, or so-called C3 glomerulopathy, will usually have C3 only or at least C3 predominance (Figure 17-22D). The histologic, IF, and ultrastructural lesions for specific diseases are detailed below, but a careful inventory of the lesions in all renal compartments—glomeruli, tubules, interstitium, and vessels—and correlation of the morphologic findings with the clinical history and the results of renal function tests and serologic studies are necessary for the proper clinicopathologic interpretation of renal biopsy specimens from patients of any age.
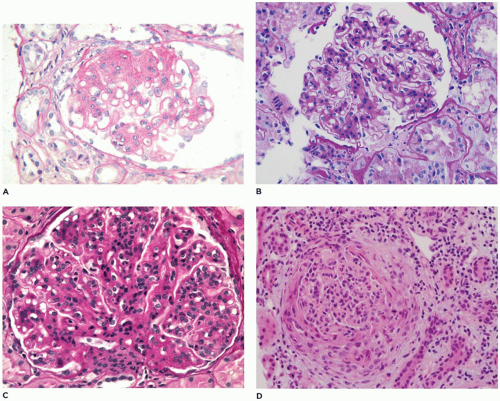 FIGURE 17-21 • Glomerular lesions observed in pediatric renal biopsy specimens. A: FSGS with the sclerotic tuft in the 11 o’clock position adherent to Bowman capsule and segmental proliferation of visceral epithelial cells at the 2 o’clock to 4 o’clock position. B: Mesangial proliferation is defined as more than three mesangial cell nuclei per peripheral mesangial focus. C: Mesangiocapillary or MPGN shows both mesangial proliferation and thickening of capillary loops. D: In CGN, a segmental, or in this case, circumferential proliferation of epithelial and inflammatory cells in Bowman space compresses the underlying glomerular tuft. (D: hematoxylineosin, A-C periodic acid-Schiff stain, original magnifications ×400.) |
9% to 40%; membranous glomerulonephritis (MGN), 1.5% to 9%; and chronic or unclassified glomerulonephritis, 6% to 17% (126). The incidence of MCD is higher in children under 6 years old at diagnosis (71% versus 24%), more frequent in males (male to female ratios of 60:40), and less often associated with hypertension or hematuria than other causes of nephrotic syndrome. A response to prednisone at 8 weeks was seen in 93% of patients with MCD, 75% with focal global glomerulosclerosis, 30% with FSGS, 56% with diffuse mesangial hypercellularity, 7% with MPGN, and none with MGN. MCD is underrepresented in more recent series, reflecting the current practice of not performing biopsies in children with nephrotic syndrome unless unresponsive or resistant to steroid therapy. However, even allowing for the changes in biopsy practice, the incidence of FSGS in children appears to be increasing in all ethnic groups, becomes more apparent in patients over 6 years of age at presentation, and appears to be more common and more aggressive in African American and possibly Japanese children (127). Familial FSGS has been attributed to mutations that alter slit-diaphragm proteins (nephrin [NPHS1], podocin [NPHS2], CD2-associated protein [CD2AP], short transient receptor potential channel 6 [TRPC6]), actin-regulating proteins (alpha-actinin-4 [ACTN4], inverted formin-2 [INF2], Rho GTPase-activating proteins 24 [ARHGAP24], Rho GDP-dissociation inhibitor 1 [ARHGDIA]), transcription factors (Wilms tumor protein [WT1], LIM homeobox transcription factor 1β [LMX1B], WSI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein [SMARCAL1]), glomerular basement membrane proteins (laminin subunit β2 [LAMB2], integrin β4 [ITGB4]), and various mitochondrial and lysosomal proteins. Many of these patients do not
present until adulthood, and there is considerable variability in the clinical severity and response to therapy, especially among heterozygotes, implying that other genes or nongenetic triggers may be involved (127,128). Altered expression or distribution of these and other podocyte proteins has been found in studies of nonfamilial FSGS suggesting that reorganization of podocyte proteins in response to injury is a key step in the pathogenesis of proteinuria (129). Gene expression profiling of isolated glomeruli from patients with FSGS, MCD, and normal controls demonstrated numerous significantly differentially regulated genes including some known to be part of the slit-diaphragm complex and previously described in dysregulated podocyte phenotype (130). FSGS, often with lesser (nonnephrotic) levels of proteinuria, is also the lesion seen in cyanotic congenital heart disease, sickle cell anemia, massive obesity, HIV, and other viral infections (parvovirus, SV40, and some cases of hepatitis C). Nephrotic syndrome in the first year of life may be associated with lesions that occur in older children but is more often caused by one of two lesions unique to this age group—congenital nephrotic syndrome (CNS) of the Finnish type (CNF) and diffuse mesangial sclerosis (DMS) that are discussed later. Medical complications of nephrotic syndrome include acute infections and thromboembolic disease related to the nephrotic state and long-term effects on bones, growth, and the cardiovascular system related to the disease and its treatment (125).
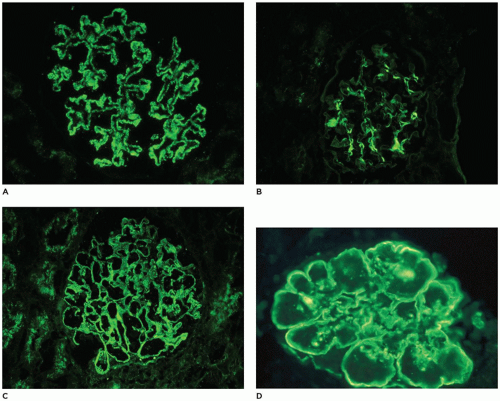 FIGURE 17-22 • Immunofluorescence patterns observed in pediatric renal biopsy specimens. A: Granular staining along the capillary loops. B: Confluent granular staining in mesangia. C: Combination of capillary and mesangial granular staining. D: Linear staining for C3 along the capillary wall and bright rings with mesangia in dense deposit disease (type II MPGN). [Fluorescein isothiocyanate-conjugated anti-IgG (A-C) or anti-C3 (D), original magnifications 400×]. |
most patients is initiated without a biopsy and 90% to 95% of patients respond within 8 weeks, although approximately 60% relapse (126). Less frequent relapse is reported in those who receive extended initial therapy, and supplemental therapies have included calcineurin inhibitors, especially cyclosporine; alkylating agents, particularly cyclophosphamide; and more recent introduction of mycophenolate mofetil (MMF) and rituximab (141). Secondary causes of MCD include NSAIDs, lymphomas, infections, and allergic reaction, and the diagnosis is suspected by clinical history.
present, but a “full house” of immunoreactants suggests lupus or another systemic disease. Mesangial deposits also suggest systemic disease. Four stages have been described in MGN: stage I, small subepithelial deposits (Figure 17-23C); stage II, larger and more numerous deposits bordered by projections of the lamina densa; stage III, incorporation of deposits into the lamina densa (Figure 17-23D); and stage IV, a thickened and irregular basement membrane without recognizable deposits. These observations provide much insight into the morphology of MGN, but they simplify the biologic complexity as the stages do not correlate with proteinuria, outcome, or clinical change and often overlap. Patients may present at any stage and may have deposits characteristic of more than one stage. Foot process retraction is typically extensive in all stages. Treatment is influenced by clinical severity; asymptomatic children with nonnephrotic proteinuria are often managed conservatively with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, while those with nephrotic syndrome receive various combinations of corticosteroids, alkylating agents, or other immunosuppressants such as calcineurin inhibitors, MMF, and rituximab (150,159).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


