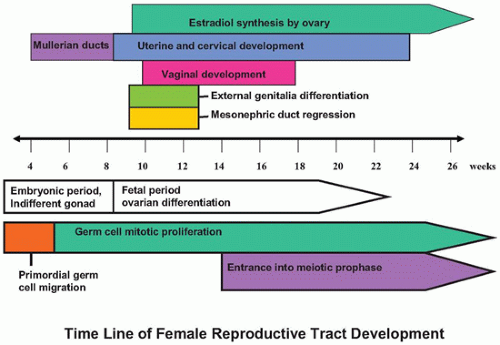
The female reproductive tract consists of the gonads, a ductal system (fallopian tubes, uterus, cervix, vagina), and the external genitalia (clitoris, labia majora, labia minora, vestibule, mons pubis). The process of sexual differentiation can be divided into various phases. Chromosomal (genetic) sex is determined by the XY or XX genotype, with the potential for abnormal deletion or addition of sex chromosomal material such as in Turner (45,X) and Klinefelter syndromes (47,XXY). Gonadal sex refers to gonadal differentiation into testis or ovary and is predominantly dependent on the expression of the sex-determining region on Y gene (SRY). SRY begins a cascade of molecular signals, resulting in male gonadal differentiation; the inhibition of these male-specific signals and the presence of several female-specific molecular signals result in female gonad differentiation. Phenotypic sex refers to the differentiation of the ductal system and of the external genitalia, a process that is regulated by production of the antimüllerian hormone (AMH) by the Sertoli cells of the testis and steroid hormone production by the testis (testosterone/dihydrotestosterone) and the ovary (estrogen/progesterone). Lastly, the assigned or adopted sex is usually determined by the chromosomal, gonadal, and phenotypic sex at birth but may be altered postnatally in a variety of disorders of sex development (DSD, see below). These processes occur predominantly in utero, with the final phenotypic changes being initiated by the onset of puberty. Previously, it was thought that sex determination involved the initiation of molecular and cellular events leading to male gonad development and that if this pathway failed, there was a default to a female gonadal phenotype. We now know that there are specific male and female molecular pathways that are needed to initiate, complete, and maintain the respective sex phenotypes as well as pathways that prevent the other sex from developing. Specific alterations in these pathways during development can result in partial male and female gonads (e.g., ovotestis) and/or ambiguous genitalia.
Primordial Germ Cells
The primordial germ cells (PGCs) first appear in the wall of the yolk sac at about 3 to 4 weeks after fertilization and migrate through the hindgut and mesonephric ridge to the genital ridge beginning about week 4 to 5, a process mediated by a number of molecular factors (
1,
2,
3,
4). Germ cells that do not reach the genital ridge are thought to undergo apoptosis, but they have also been proposed as the cell of origin of extragonadal germ cell tumors. PGCs are bipotential, and complete differentiation into oogonia or prospermatogonia depends upon the local environment. The initial events in testicular and ovarian differentiation are independent of the presence or genotype of PGCs in the gonad. However, completion of appropriate ovarian development depends upon the presence of meiotic germ cells. In the absence of meiotic germ cells, the ovarian structure degenerates leaving streak ovaries (
5,
6). Germ cell sexual dimorphism begins at about 11 to 12 weeks of gestation. Male PGCs (prospermatogonia) enter mitotic arrest, whereas ovarian PGCs (oogonia) enter the first meiotic prophase and then arrest. We now know that retinoic acid produced locally by the ovary induces PGC entry into meiosis beginning in the medulla and slowly radiating out to the cortex. Male germ cells produce the retinoic acid-degrading enzyme CYP26B1 via paracrine FGF9 signaling from male support cells to prevent entry into meiosis.
Early Gonadal Development
The gonadal ridge develops at the ventromedial aspect of the mesonephros by a proliferation of mesodermal cells and thickening of the overlying coelomic epithelium at 4 to 5 weeks’ gestation. Between 4.5 and 6 weeks, the indifferent gonad is indistinguishable as male or female (
Figure 18-1). A number of critical genes have been identified in regulating normal genital ridge development including SF1, WT1, LHX9, CITED2, IGF signaling, and potentially EMX2 and PBX1. In humans and mice, the Wilms tumor gene isoform WT1-KTS as well as the steroidogenic factor 1 (previously referred to as SF1 and now officially designated NR5A1) genes are important in maintaining the indifferent gonad. Mutations in SF1 can lead to adrenal-gonadal failure, mostly affecting male gonad development, as maintenance of SF1 expression is also critical in testis development. The WT-KTS isoform is required for cell survival and proliferation within the bipotential gonad in both males and females (
5). WT1+KTS isoform is critical later for male development, and the ratio of +KTS/-KTS seems critical for appropriate gonadal differentiation. Mutations in WT1 are associated with three syndromes characterized by gonadal dysgenesis (Wilms tumor/aniridia/gonadal dysgenesis/retardation—WAGR [OMIM 194072], Denys-Drash [OMIM 194080], and Frasier [OMIM 136680]) (see www.omim.org or www.ncbi.nlm.nih.gov/omim). The phenotype of affected individuals includes genital and kidney defects as well as an increased risk for the development of Wilms tumors. Insulin signaling occurs via IGF1 and IGF2 and is critical for cell proliferation during early gonadal development. In the absence of proper insulin signaling, the gonads can be very small or absent. At about 7 weeks’ gestation, in XY embryos, a Y-linked genetic switch, SRY, is expressed by pre-Sertoli cells (
1,
2,
3,
4,
5). The expression of SRY is necessary and sufficient to trigger male testis development; however, both the timing and the level of SRY expression are critical for normal testis development. In an XY embryo, the lack of SRY results in ovarian development. The SRY protein contains a highly conserved DNA-binding domain that allows specific genes to be turned on or off. The primary target is SOX9, which is thought to be the key Sertoli cell differentiation factor. Both SRY and SF1 bind upstream of SOX9 to initiate transcription. Once SOX9 transcription is initiated, there is a feedforward loop whereby SOX9 itself replaces SRY and binds upstream to maintain its own transcription. SOX9 also induces FGF9, which stimulates more SOX9 production. SOX9 is essential for production of the first male-specific cell type, the Sertoli cell. Mutation in SOX9 leads to the human dwarfism syndrome camptomelic dysplasia (OMIM 114290), which is often associated with XY sex reversal (
7). Duplication of SOX9 causes XX femaletomale sex reversal. SF1 expression in the developing testis regulates expression of several male-specific genes, including AMH (see
Chapter 28). It has been recently discovered that SOX9 activation results in increased prostaglandin D2 (PGD2) production, which is essential for maintenance of the testis phenotype by facilitating Sox9 transportation into the nucleus. In animal models, there are well-documented examples of both male and female maintenance factors (e.g., PGD2), which if disrupted even postnatally can result in gonadal transdifferentiation to the alternative gonad. Leydig cells are derived from mesenchymal precursors via desert hedgehog (DHH) signaling (
1,
2,
3,
4).
Ovarian Differentiation
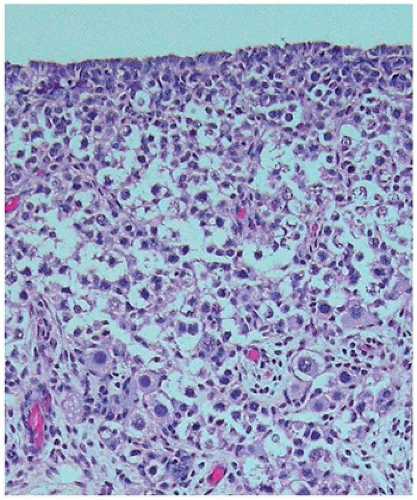
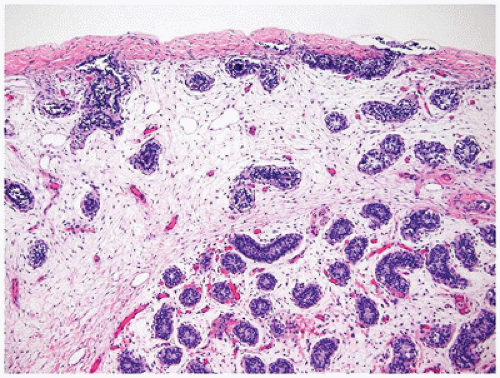
Ovarian development is thought to be characterized by active ovarian determination pathways as well as by inhibition of testis-specific pathways. In addition, it is now clear that once established, maintenance of the ovarian phenotype occurs via FOXL2 and estrogen receptor alpha and beta signaling. The early formation of the ovary can be thought of as involving three major processes: (a) entrance of oogonia into prophase of meiosis I to form primary oocytes, (b) granulosa cells surrounding these primary oocytes to form primordial follicles, and (c) differentiation of the steroid-producing theca cells. By 6 weeks of development, primordial sex cords form. The ovary can be identified at 7 to 8 weeks’ gestation by the absence of true testicular cords and possessing PGC nests. After their arrival in the gonad, germ cells in the female continue to undergo active mitotic cell division, a process that diminishingly continues into the third trimester at least in the cortex. It is estimated that approximately 3 to 4 million germ cells are present in each ovary by 20 weeks’ gestation, and then the number decreases to about 0.5 to 1 million at term. The mechanism leading to the loss of oocytes remains poorly understood. At approximately 12 weeks’ gestation, the first germ cells begin to enter into meiosis via retinoic acid signaling, a process first seen close to the medullary region (
Figure 18-2). On entering meiosis, the primary oocyte will arrest at the diplotene stage of the first meiotic prophase and become enclosed by follicular cells to form primordial follicles. Follicular or granulosa cells are in direct contact with the germ cells and are thought to
play a role in regulating continued meiotic arrest in the germ cells. Oogenesis is a gradual process that is generally complete by the third trimester, and there is no further increase in the number of primary oocytes thereafter (
Figure 18-3). However, in the mouse germ or stem cell, replication may continue into adulthood. In addition to the germ cells, the ovary is populated by stromal cells that continue to remodel during the early first trimester, resulting in the formation of the ovarian cortex and the medulla. During the second trimester, a subepithelial collagenous connective tissue layer develops within the ovarian cortex beneath the basement membrane. The interstitial (thecal) cells, of unknown origin, can be detected during the first half of the second trimester, although estrogen production may begin as early as 8 to 10 weeks’ gestation. The importance of the production of estrogen by the developing fetus remains somewhat controversial with some evidence that significant ovarian estrogen production does not occur until following birth (
6). The histology of the developing ovary has been previously described in detail (
8).
While many of the molecular details of testis development have been clearly established, the details regulating molecular ovarian development are now coming to light. The NR0B1 gene, previously designated DAX-1, is a dosagesensitive gene locus on the X chromosome that encodes an orphan nuclear receptor protein. This gene was initially thought to be a specific ovary-determining gene but has since been shown to be more essential for normal testicular development and not required for normal ovarian development. NR0B1 is expressed in the normally developing ovary but turned off in the developing testis (
5,
6). Mutations of NR0B1 in XY males lead to hypogonadotropic hypogonadism with primary testicular defects and are associated with adrenal insufficiency (X-linked adrenal hypoplasia congenita). Loss of function of NR0B1 in XX females does not alter normal ovarian development. Overexpression of NR0B1, however, leads to ovarian development in males even if SRY is expressed. NR0B1 therefore appears to be more important in normal testis development. Unlike the SRY gene and testis development, there does not appear to be a single ovarian determining gene. The canonical WNT signaling pathway is specifically activated in XX gonads and diminished in XY gonads. WNT4 is expressed in both early gonads but is then up-regulated in ovarian somatic cells, and down-regulated in the testis. WNT4 expression seems critical for some portions of ovarian development, but not all. Constitutive activation of beta-catenin in XY gonads results in sex reversal with a XY female gonad. Without WNT4 expression in XX individuals, male-specific changes occur that include the presence of steroid-producing cells within the ovary, persistence of the wolffian ducts, loss of the müllerian ducts, and the development of a male-specific coelomic blood vessel to the ovary. In the absence of WNT4, germ cells can still enter meiosis, but there is massive apoptosis of the germ cells prior to birth. Another recently identified gene that
is involved in activating the WNT/beta-catenin pathway is a pro-ovarian gene, RSPO1 (R-spondin-1). Homozygous mutation in RSPO1 results in female-to-male sex reversal in XX individuals. It would seem that activation of the canonical WNT signaling pathways is required to fully suppress the testis-determining pathway. WNT4 signaling appears to be necessary for (a) normal development of the müllerian duct, (b) suppression of the Sertoli cell lineage in the developing ovary, and (c) oocyte maintenance (
1,
2,
3,
4,
6). Another gene proposed to be involved in ovary-specific development is FOXL2. FOXL2 seems to be critical for maintenance of the ovarian phenotype by repressing SOX9 expression and therefore preventing testicular differentiation. Mutations in the forkhead transcription factor 2 (FOXL2) gene in humans are associated with eyelid defects and premature ovarian failure (blepharophimosis, ptosis, and epicanthus inversus syndrome—BPES [OMIM 110100]). FOXL2 is expressed by pregranulosa cells (
9). While estrogen secretion
in utero does not appear to be important for sex determination, estrogen signaling does appear to be critical in the maintenance of the maturing ovary later during puberty. Blocking estrogen signaling in puberty results in transdifferentiation of the ovary and expression of Sertoli cell markers. Estrogen signaling appears to be critical in maintaining the mature ovarian structure by suppressing testicular development during early adult development. Estrogen signaling seems to antagonize activation of the transcriptional enhancer site for SOX9, thereby helping prevent SOX9 expression. It would seem that the gonadal phenotype is maintained early and throughout life by repression of the molecular pathways of the opposite sex. This helps to explain the wide variety of gonadal and genital anomalies including ovotestis.
The ovary differs from the testis in that the presence of germ cells is essential for normal ovarian development. In the developing testis, the male germ cells do not play a significant role in the structural development of the organ. Female germ cells appear to follow an intrinsic clock to enter the first meiosis and arrest prior to completion. Once female germ cells have entered meiosis, they have committed to the oocyte fate. During fetal development, the oocyte becomes surrounded by a single layer of granulosa cells to form the primordial follicle. Early primordial follicle formation and maintenance involve several complex molecular pathways acting sequentially and together (reviewed in (
4,
10,
11)). Maturation of these follicles can proceed under hormonal signaling, especially during the third trimester of pregnancy.
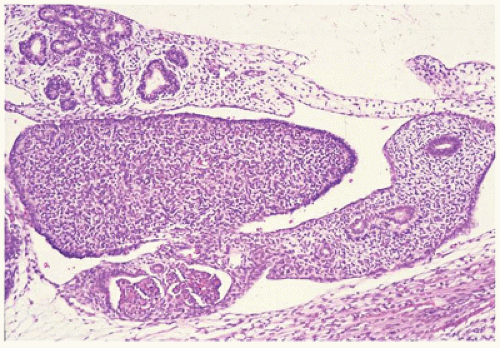
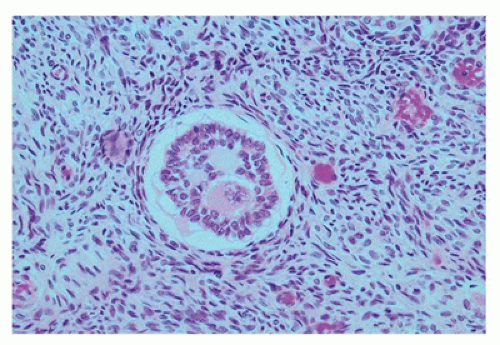
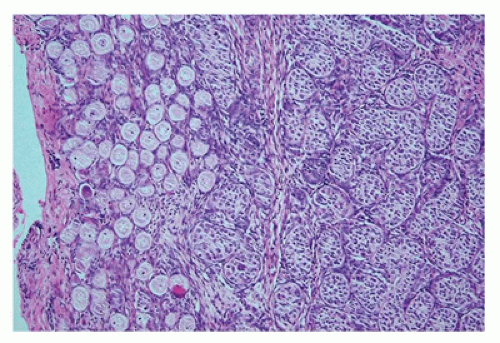
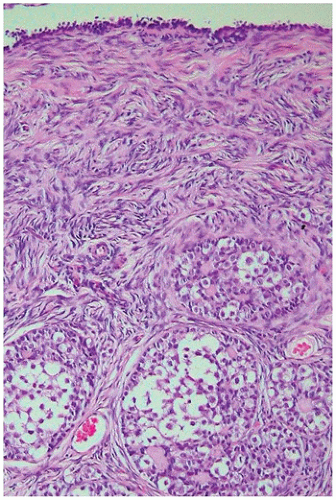
At birth, the ovary is tan, flat, and elongated and measures about 1.3 × 0.5 × 0.3 cm. and weighs less than 0.3 g. Before birth, some primordial follicles can develop further. The ovum enlarges, and the surrounding follicular cells become more cuboidal to columnar and thereby form a primary follicle. This may be followed by stratification of the granulosa cells and increased granulosa cell proliferation, resulting in a preantral follicle (
Figure 18-4). The graafian follicle demonstrates a cavity within the granulosa cell layer. The granulosa cells in these follicles have scant cytoplasm and often surround cavities filled with deeply eosinophilic material, known as
Call-Exner bodies that may result in microscopic structures resembling gonadoblastoma or annular tubulelike profiles. These likely represent abnormal folliculogenesis. Thecal cells, which differentiate from the stromal cells at the periphery of developing follicles, may be seen. Throughout childhood, the ovaries enlarge to reach the size and the shape of an adult ovary (4 × 2 × 1 cm, 5 to 8 g). During the prepubertal period, the number of oocytes and primordial follicles continues to decrease, and the amount of ovarian stroma increases. Like the testicular Leydig cells, ovarian hilus cells disappear during childhood and reappear during puberty.
The Ductal System
The development of the urinary and reproductive systems is highly interdependent, which explains the high incidence of coexisting genital and urinary tract anomalies in several syndromes. Early in development (approximately week 4), paired mesonephric (wolffian) ducts arise from the intermediate mesoderm and nephrogenic cord (
Figure 18-5). In the female embryo, the mesonephric duct is needed for induction of paramesonephric (müllerian) duct development from invaginations of the coelomic epithelium. The mesonephric and the paramesonephric ducts are enclosed in a peritoneal fold that gives rise to the broad ligament of the uterus. In the male embryo, AMH produced by fetal Sertoli cells leads to regression of the ipsilateral paramesonephric duct between weeks 8 and 10. AMH is proposed to have multiple functions in women (
12). After the sensitivity of the paramesonephric ducts to AMH has disappeared, the ovarian granulosa cells begin to produce AMH with increasing levels that peak at about 10 years of age. AMH is thought to maintain meiotic arrest in the oocyte of the developing follicle in prepubertal females (
10,
11).
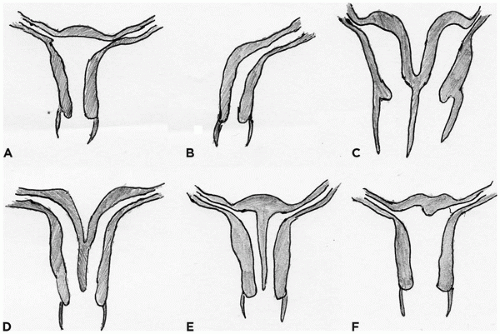
In the female embryo, the paramesonephric ducts fuse caudally before reaching the urogenital sinus, a process that is completed by week 10 (
Figure 18-5). The unfused paramesonephric ducts become fallopian tubes and the fused portions the uterus and the upper vagina. The distal tip of the müllerian duct abuts the posterior wall of the urogenital sinus
within a patch of mesoderm. This point is the future site of the hymenal membrane. The patch of mesodermal urogenital sinus epithelium begins to proliferate, forming a column of squamous cells called the
vaginal plate that eventually gives rise to the vaginal epithelium. The vaginal plate and the müllerian duct become patent by canalization early in the second trimester (by week 18). Mesonephric ducts in the female embryo begin to regress if not stimulated by testosterone by about week 10; however, mesonephric remnants in the broad ligament and lateral wall of the uterus and vagina can persist as Gartner ducts. By 13 weeks’ gestation, the body (corpus) of the uterus and the cervix begin to be distinguished. In the fetus and the newborn, the cervix is twice as long as the corpus, whereas in the adult, the corpus is about two times longer than the cervix. At birth, the cervix and uterus together measure about 4 cm in length. The effects of maternal hormones (estrogens and progestins) result in a proliferative-to-weakly secretory endometrium at birth and cervical squamous cell maturation. These changes rapidly disappear after birth. During childhood, the endometrium is usually thin, with inactive glands in a spindled inactive stroma. The uterus reaches a plateau of growth in the 2nd year of life, until the premenarchal uterine growth increase. The final adult (nulliparous) uterus measures 7 to 8 cm in longest dimension and weighs between 40 and 80 g. The final maturation of the female reproductive tract is at the beginning of uterine bleeding (menarche), which occurs between 11 and 15 years of age. The early menstrual cycles are often anovulatory and can result in disordered proliferative endometrium. By mid-adolescence, regular menstrual cycles should be occurring, along with the monthly histologic changes that are well described for the adult female reproductive tract.
A number of genes have been reported as important in regulating normal internal female genital development. WNT signaling is essential for normal müllerian duct development. Spatial and temporal expression patterns of members of the HOXA gene locus are essential for normal development of the fallopian tubes (HOXA9), uterus (HOXA10), cervix (HOXA11), and upper vagina (HOXA13). In mice, many transcription factors have been implicated in normal müllerian duct formation including Lim1, Pax2, Emx2, several Wnt members (Wnt4, Wnt5a, Wnt7a, Wnt7b, Wnt11), and members of the retinoic acid receptor family (Rar-alpha, beta, and gamma) (
13). Estrogen receptors alpha and beta are highly expressed in the female internal genital tract and are important for attaining the final functional phenotype especially for the uterus. Knocking out both ERa and ERb results in a hypoplastic uterus (reviewed in (
13,
14,
15,
16)).
Female External Genitalia
The external female genitalia begin to form during the 4th week of embryonic development. The genital tubercle forms ventral to the cloacal plate as two stromal elevations of the ectoderm. Lateral to the cloacal plate on each side, two parallel folds develop, which will give rise to the labia majora and the minora. As the labioscrotal folds extend cranially around the genital tubercle, they fuse and become the mons pubis (
17,
18). Sonic hedgehog signaling is an important regulator of genital tubercle formation, as well as other downstream genes including WNT5a and FGF8. The role of estrogen in the development of the entire female reproductive tract is unknown. Androgen production in the male is a critical regulator of penis and scrotum formation. If a female fetus is exposed to elevated androgens before 10 to 12 weeks of gestation, the external genitalia may become ambiguous or resemble a phenotypic male and the vagina will often open into the membranous portion of the urethra. If androgens become elevated after week 20, the only effect will be an enlarged clitoris. Similarly, androgen insensitivity due to mutations in the androgen receptor in XY males results in female external genitalia (
16,
17).
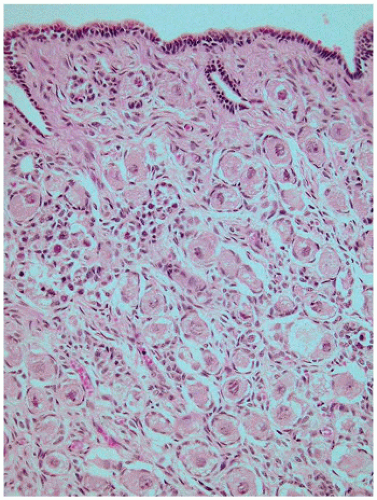
The entire vulva, with the exception of the vestibule, is lined by keratinized, stratified squamous epithelium. The vestibule and the vagina are lined by nonkeratinizing squamous epithelium, which becomes glycogenated in women of reproductive years, and should not be confused with koilocytotic change. The vaginal vestibule contains the orifices of the paraurethral (Skene) glands, the major (Bartholin) and minor vestibular glands, and the urethral meatus. The paired paraurethral glands are located on either side of the urethral meatus, are composed of pseudostratified mucus-secreting columnar epithelium, and are drained by ducts lined by transitional epithelium. The vestibular glands contain acini composed of simple columnar, mucus-secreting epithelium. The major vestibular (Bartholin) glands are drained by ducts lined proximally by mucus-secreting epithelium epithelium and more distally by transitional epithelium with terminal lining by squamous epithelium on their exit just external to the hymenal ring. The minor vestibular glands are located close to the surface and ring the vestibule.