Chapter 11 THE ENDOCRINE SYSTEM
Introduction
 Endocrine diseases affect the overall intermediary metabolism. Lack of thyroid hormones leads to a slowing down of the body’s metabolism, whereas hyperthyroidism leads to hypermetabolism.
Endocrine diseases affect the overall intermediary metabolism. Lack of thyroid hormones leads to a slowing down of the body’s metabolism, whereas hyperthyroidism leads to hypermetabolism.




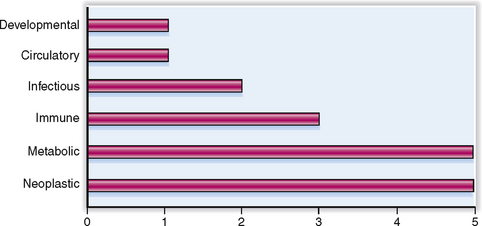
In this chapter we discuss only the diseases affecting the major endocrine organs: pituitary, thyroid, parathyroids, and adrenal glands. The cells forming the dispersed endocrine system of the gastrointestinal and respiratory tract are not discussed. Diabetes mellitus, the most common and clinically the most important endocrine disease, was discussed in the Chapter 10. The relative clinical significance of various endocrine diseases is presented graphically in Figure 11-1.
Anatomy and Physiology
Adenohypophysis Anterior portion of the pituitary gland comprising the pars tuberalis, pars distalis, and pars intermedia. In standard histologic sections it is composed of three types of cells: acidophilic, basophilic, and chromophobic cells. These cells secrete polypeptide hormones and can be classified functionally or immunohistochemically into six groups: somatotrophs (growth hormone-secreting), lactotrophs (prolactin-secreting), thyrotrophs (thyroid-stimulating hormone-secreting), corticotrophs (adrenocorticotropic hormone-secreting), gonadotrophs (follicle-stimulating hormone and luteinizing hormone-secreting), and hormonally inactive cells (“null” cells, or stem cells).
Adrenal cortex Outer part of the adrenal glands. It comprises three layers: zona glomerulosa composed of mineralocorticosteroid-secreting cells, zona fasciculata composed of glucocorticoid-secreting cells, and zona reticularis composed of androgen-secreting cells.
Adrenal glands Paired endocrine glands located retroperitoneally just above the kidneys. They consist of two parts: the outer, known as the cortex, and the inner, known as the medulla.
Adrenal medulla Inner part of the adrenal glands. It is composed of catecholamine-secreting chromaffin cells. These cells develop from the neural crest cells, which have migrated to the retroperitoneal area during fetal development. The same neural crest cells give rise to paraganglia and autonomic ganglia, and thus tumors like the pheochromocytoma and neuroblastoma may originate either from adrenal or extra-adrenal sites.
Aldosterone Mineralocorticosteroid produced by the zona glomerulosa of the adrenal cortex. Its secretion from the cells of the zona glomerulosa is controlled mainly by the renin–angiotensin system. In the kidneys aldosterone promotes reabsorption of sodium and bicarbonate and the excretion of potassium and hydrogen ions. Secondary retention of water may lead to arterial hypertension.
Androgens Male sexual hormones produced by the Leydig cells of the testis and adrenal cortical cells in the zona reticularis. The most important androgens are testosterone, androstenedione, dehydroepiandrosterone (DHEA), and dehydroepiandrosterone sulfate (DHEAS).
Antidiuretic hormone (ADH, vasopressin) Octapeptide hormone synthesized by neuronal cells in the hypothalamic nuclei and stored in the posterior lobe of the pituitary. Its synthesis is regulated by the osmolality of the plasma. Antidiuretic hormone stimulates the contraction of smooth muscles. Its effects on arterioles raise the blood pressure, and its effects on the intestines promote peristalsis. The antidiuretic effect results from its action on the distal tubules of the kidney, where it promotes the resorption of water.
Catecholamines Group of biogenic amines, the most important of which are dopamine, epinephrine, and norepinephrine. Catecholamines found in the blood are derived from the adrenal medulla. Epinephrine and norepinephrine are also found in the central nervous system and peripheral nerves. Catecholamines have many sympathomimetic effects (“fight-or-flight” reaction in stress), and raise the blood pressure.
Corticosteroids Group of 21-carbon steroids produced from progesterone by adrenal cortical cells. According to their function they are classified as mineralocorticoids or glucocorticoids.
Corticotropin (adrenocorticotropic hormone, ACTH) Glycopeptide produced in the anterior lobe of the pituitary under the control of the hypothalamic corticotropin-releasing hormone (CRH). It stimulates the adrenal cortical cells and is also excreted in large amounts during stress.
Follicle-stimulating hormone (FSH) Gonadotropic glycopeptide produced in the anterior lobe of the pituitary under the control of the hypothalamic FSH-releasing hormone. It stimulates the growth of ovarian follicles and secretion of estrogen. In males it stimulates spermatogenesis.
Glucocorticoids Steroid hormones produced by the zona fasciculata of the adrenal cortex. The most important glucocorticoids are cortisol (hydrocortisone), cortisone, and corticosterone. Their secretion is under the control of ACTH and is increased in stress. They affect the metabolism of carbohydrates, lipids, and proteins.
Growth hormone (GH) Polypeptide hormone secreted by cells of the anterior pituitary. It stimulates the liver to produce somatomedins, which promote skeletal growth. Growth hormone also acts on the intermediary metabolism of carbohydrates, lipids, and proteins.
Luteinizing hormone (LH) Gonadotropic glycoprotein produced by the anterior lobe of the pituitary under the control of the hypothalamic LH-releasing hormone. It promotes ovulation and stimulates the secretion of progesterone from the corpus luteum in women. In men it acts on Leydig cells, stimulating the production of androgen.
Neurohypophysis Posterior part of the pituitary composed of cytoplasmic extensions of neural cells in several hypothalamic nuclei. These cells secrete antidiuretic hormone (ADH, vasopressin) and oxytocin.
Oxytocin Nonapeptide synthesized by the cells of the paraventricular nucleus of the hypothalamus and stored in the posterior lobe of the pituitary. It is released on stimulation of cholinergic fibers in the areolar area of the nipple during suckling. It causes uterine contractions during delivery and milk release during lactation.
Parathormone (parathyroid hormone, PTH) Polypeptide hormone produced by the parathyroid glands. It stimulates hypercalcemia by promoting calcium release from the bone, calcium absorption in the intestine, and renal calcium reabsorption.
Parathyroid glands Four small endocrine glands located on the posterior side of the thyroid. They secrete parathyroid hormone, a polypeptide hormone essential for the metabolism of calcium and phosphorus.
Pituitary Endocrine gland located in the sella turcica and attached by a stalk to the hypothalamus. It consists of an anterior and intermediate part forming the adenohypophysis and a posterior part forming the neurohypophysis.
Prolactin Polypeptide hormone secreted by the anterior lobe of the pituitary. It plays an important role in the development of the mammary glands during pregnancy and the production of milk during lactation. It also suppresses LH secretion causing anovulation and amenorrhea.
Thyroglobulin High-molecular-weight iodinated glycoprotein produced by thyroid follicular cells and stored in the colloid. It is taken up by follicular cells and transformed into thyroxine and triiodothyronine.
Thyroid Bilobed endocrine gland located on the neck anterior to the trachea. It is composed of thyroid hormone-secreting cells arranged into follicles, and scattered C cells secreting calcitonin.
Thyroxine (tetraiodothyronine, T4) Principal hormone produced by the thyroid. It is partially diodinated into a more active form, triiodothyronine (T3). Both T4 and T3 stimulate metabolism and act on most major organs in the body.
Clinical Signs and Symptoms
Amenorrhea Lack of menstrual bleeding, usually resulting from hormonal disturbances involving the pituitary gland or the ovary. If the menstrual bleeding never occurs at the time of puberty it is called primary amenorrhea. Cessation of menstruation in a woman who was previously menstruating normally is called secondary amenorrhea.
Dwarfism (nanosomia) Condition in which the body is considerably smaller than normal. It may have many causes, including pituitary or thyroid insufficiency in infancy and childhood.
Exophthalmos (proptosis) Abnormal bulging of the eyes, typically seen in Graves’ disease.
Galactorrhea Abnormal production of milk unrelated to pregnancy and lactation, or an uncontrolled flow of milk during lactation.
Goiter Diffuse, usually nodular enlargement of the thyroid caused by a variety of conditions, such as thyroiditis, iodine deficiency, or hormonal disturbances including thyroid enlargement.
Hirsutism Condition characterized by excessive hairiness. In women it is characterized by hair growth in a male pattern (e.g., face and chest).
Myxedema Form of nonpitting chronic edema predominantly involving the face and causing swelling of the lips and nose and deepening of facial furrows.
Osteomalacia Form of osteopenia (reduced bone mass) characterized by inadequate mineralization of the osteoid matrix.
Osteoporosis Form of osteopenia (reduced bone mass) characterized by a loss of mineralized bone and a tendency to fractures.
Endocrine Diseases
Acromegaly and gigantism Syndrome caused by an excess of growth hormone (GH), usually produced by a pituitary adenoma. In children and adolescents excess GH leads to tall stature (gigantism). Acromegaly (i.e., an enlargement of the acral parts—feet, hands, nose, and jaw) is found in adults. Additional findings include hypertension, cardiomegaly, insulin resistance, and metabolic disturbances such as hyperglycemia.
Addison’s disease Adrenal cortical insufficiency due to the destruction of the adrenals by autoimmune diseases, infections (e.g., tuberculosis), or metastatic tumors. It is characterized by weight loss, weakness, hypoglycemia, hypotension and hyperpigmentation, and laboratory findings indicative of a mineralocorticoid and glucocorticoid deficiency.
Amenorrhea–galactorrhea syndrome Syndrome related to an excess of prolactin, usually secreted by a pituitary adenoma. Clinically it manifests with spontaneous milk secretion and cessation of the menstrual cycle.
Conn’s syndrome (primary hyperaldosteronism) Syndrome caused by an excess of aldosterone, usually produced by a cortical adenoma. It is characterized by hypertension, metabolic alkalosis, polyuria, and potassium wasting. Laboratory findings include hyperaldosteronism, low renin, and low serum potassium. Secondary hyperaldosteronism is a consequence of renal diseases leading to hypersecretion of renin.
Cushing’s syndrome Syndrome caused by an excess of glucocorticoids, produced by the pathologically altered or overstimulated adrenal cortical cells, or due to an iatrogenic intake of steroids. It is clinically characterized by truncal obesity, moon facies, buffalo hump, contrasted with thin extremities affected by muscle wasting. Osteoporosis, hypertension, skin atrophy, and reduced resistance to infections are also present. Laboratory findings include excess glucocorticoids in the blood and urine and corresponding changes involving minerals and glucose. Adrenocorticotropic hormone is usually suppressed, except in patients who have Cushing’s disease due to corticotropic adenoma of the pituitary.
Diabetes insipidus Syndrome related to ADH deficiency (central diabetes insipidus) or an inability of the kidneys to respond to proper ADH stimulation (renal diabetes insipidus). In either case the kidneys cannot retain water, resulting in polyuria and dehydration. Despite polydipsia, serum osmolality is increased, as is the specific gravity of the urine. Hypernatremia and decreased osmolality are also features.
Graves’ disease Autoimmune disease characterized by overstimulation of the thyroid with antibodies to thyroid-stimulating hormone (TSH) receptors. It is clinically characterized by symmetric enlargement of the thyroid, hyperthyroidism, and exophthalmos.
Hyperparathyroidism Syndrome caused by an excess of parathyroid hormones. It may be primary (most often due to parathyroid adenoma) or secondary (due to parathyroid hyperplasia in renal insufficiency). Clinically it manifests with hypercalcemia, loss of bone substance (osteitis fibrosa cystica), renal stones, gastrointestinal disturbances, and neurologic symptoms.
Hyperthyroidism Syndrome caused by an excess of thyroid hormone, most often related to autoimmune disorders (Graves’ disease), or functionally active thyroid tumors. Clinically it manifests with signs of hypermetabolism, such as tachycardia, sweating, and nervousness.
Hypoparathyroidism Syndrome caused by inadequate parathyroid hormone production, most often due to surgical removal of all four parathyroid glands. The lack of PTH leads to hypocalcemia producing signs of neuromuscular irritability (e.g., tetanic spasms, Chvostek’s sign, Trousseau’s sign), electrocardiographic (ECG) changes.
Hypothyroidism Syndrome caused by a lack of thyroid hormones. In infants it is most often caused by congenital thyroid aplasia and is associated with growth retardation and mental retardation (cretinism). In adults it is usually due to Hashimoto’s thyroiditis or surgical resection of the thyroid. Laboratory findings include low T4 and T3 and high thyroid-stimulating hormone (TSH). Clinical findings include myxedema and slowed down metabolism and major vital function (e.g., bradycardia, constipation, drowsiness).
Infertility Inability to conceive a child or become pregnant.
Pheochromocytoma Catecholamine-secreting tumor of the adrenal medulla. Catecholamines released from the tumor cause paroxysmal hypertension and other signs of adrenergic overstimulation, such as excessive sweating, palpitations, and headaches. Excessive epinephrine and norepinephrine can be detected in the blood. Metabolized catecholamines are excreted as vanillylmandelic acid (VMA) in the urine.
Pituitary cachexia Syndrome related to a lack of pituitary hormones, typically caused by tumors of the hypothalamic area or as a result of surgical resection or trauma. Clinically it presents with wasting and weakness and signs of hypothyroidism, adrenal cortical insufficiency, and gonadal insufficiency. Laboratory findings include hypoglycemia, hyponatremia, and reduced levels of thyroid and steroid hormones. Sheehan’s syndrome is a form of panhypopituitarism found in the postpartum period. It is related to hemorrhagic infarction of the pituitary.
Normal Structure and Function
ANATOMY AND HISTOLOGY
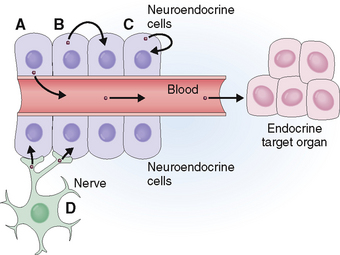
Based on the mode of hormone secretion (Fig. 11-2), endocrine and neuroendocrine cells can be classified into four categories:
 Endocrine cells. These cells secrete hormones into the blood, which carries them to a distant target organ. Typical endocrine cells are thyroid, parathyroid, and adrenal cortical cells.
Endocrine cells. These cells secrete hormones into the blood, which carries them to a distant target organ. Typical endocrine cells are thyroid, parathyroid, and adrenal cortical cells.


 Neurocrine cells. These cells have all the major features of nerve cells, but secrete hormones that act on other non-neural cells. For example, neural cells in the hypothalamus secrete releasing factors that stimulate the hormone production of pituitary cells.
Neurocrine cells. These cells have all the major features of nerve cells, but secrete hormones that act on other non-neural cells. For example, neural cells in the hypothalamus secrete releasing factors that stimulate the hormone production of pituitary cells.
PHYSIOLOGY
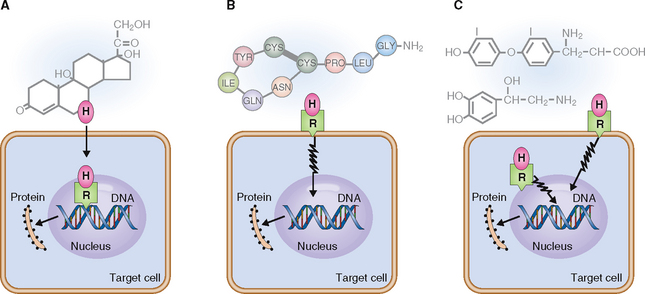
Biochemically hormones can be classified as steroids, polypeptides, and modified amino acids (biogenic amines). The main features of these hormones are briefly reviewed here and summarized in Table 11-1.

The multistep synthesis of the polycyclic backbone begins with the conversion of cholesterol into pregnenolone. This basic structure is then modified by isomerization or oxidation, leading to the formation of estrogens, progestogens, androgens, mineralocorticoids, and glucocorticoids. Steroid hormones are not water-soluble and must therefore be transported in the blood bound to transport proteins. They can enter target cells and bind to intracellular receptors, which transport them into the nucleus (Fig. 11-3). In the nucleus they bind to the hormone-response elements of the gene, stimulating protein synthesis.
Clinical and Laboratory Evaluation of Endocrine Diseases
FAMILY AND PERSONAL HISTORY
In the work-up of suspected cases of endocrine disease, begin by taking a family and personal history. Some of the risk factors that can be identified in this interview are listed in Table 11-2.
Table 11-2 Risk Factors for Some Endocrine Disorders
| TYPE OF RISK FACTOR | SPECIFIC DISEASES—RISK FACTOR ASSOCIATIONS |
| Hereditary factors | Multiple endocrine neoplasia syndromes: Medullary carcinoma of thyroid and pheochromocytoma (hypertension) |
| Nutritional factors | |
| Diseases involving other organs | End-stage kidney disease: Secondary hyperparathyroidism |
| Other endocrine diseases | ACTH-secreting pituitary adenoma: Cushing’s disease |
| Infection | Meningococcal sepsis: Acute adrenal insufficiency of Waterhouse-Friderichsen syndrome |
| Autoimmunity | Multiorgan autoimmune diseases: Hashimoto’s thyroiditis-related hypothyroidism |
| Medical and surgical procedures | Neck surgery: Hypoparathyroidism |
| Drugs | |
| External mechanical factors | Brain trauma: Hypopituitarism |
ACTH, adrenocorticotropic hormone.
PHYSICAL EXAMINATION
Symptoms and signs of endocrine disorders are often nonspecific and not readily recognizable. In some patients the endocrine disorders, such as gigantism, predominate the clinical picture, but obvious and telltale symptomatology of this sort is uncommon. Some diseases affect endocrine organs but cause few if any endocrine disturbances. For example goiter is readily detected by inspection or palpation of the neck, but it is often not associated with any disturbances in thyroid function. Many endocrine diseases are asymptomatic and are discovered only with laboratory testing.
The following is a list of some signs and symptoms of possible endocrine disease:
 Easy fatigability. Fatigue is a common symptom of many endocrine disorders, most notably hypothyroidism, hyperthyroidism, and adrenal insufficiency.
Easy fatigability. Fatigue is a common symptom of many endocrine disorders, most notably hypothyroidism, hyperthyroidism, and adrenal insufficiency.
 Weight loss. Weight loss may be associated with a loss of appetite in bulimia and anorexia nervosa. Pituitary and adrenal insufficiency cause marked wasting and cachexia.
Weight loss. Weight loss may be associated with a loss of appetite in bulimia and anorexia nervosa. Pituitary and adrenal insufficiency cause marked wasting and cachexia.













LABORATORY STUDIES
 Measure hormones in blood. For example, the concentration of thyroxine (T4) can be measured in the blood. To improve the precision of this test it is customary to also measure the concentration of T3 (the more active form of T4) and thyroid-stimulating hormone (TSH). Analyzing these three hormones in most instances determines whether hypothyroidism or hyperthyroidism is present. Furthermore, these tests can indicate whether the disease is due to a pituitary or a thyroid disorder.
Measure hormones in blood. For example, the concentration of thyroxine (T4) can be measured in the blood. To improve the precision of this test it is customary to also measure the concentration of T3 (the more active form of T4) and thyroid-stimulating hormone (TSH). Analyzing these three hormones in most instances determines whether hypothyroidism or hyperthyroidism is present. Furthermore, these tests can indicate whether the disease is due to a pituitary or a thyroid disorder.


 Use of stimulation tests. These tests are used when hormonal insufficiency is suggested, usually after initial studies have shown that the levels of a specific hormone are low. Typical stimulation tests are listed in Table 11-3.
Use of stimulation tests. These tests are used when hormonal insufficiency is suggested, usually after initial studies have shown that the levels of a specific hormone are low. Typical stimulation tests are listed in Table 11-3.
 Use of suppression tests. These tests are used when a hyperfunction of an endocrine organ is suspected. Typical suppression tests are listed in Table 11-4.
Use of suppression tests. These tests are used when a hyperfunction of an endocrine organ is suspected. Typical suppression tests are listed in Table 11-4.
Table 11-3 Endocrine Stimulation Tests
| HORMONE DEFICIENCY | TEST | EXPLANATION |
| Growth hormone | L-Dopa, arginin, or insulin stimulation test | A lack of response → hypothalamic or pituitary disorder |
| Gonadotropins (FSH and LH) | GnRH stimulation | GnRH increases FSH and LH; lack of response → hypothalamic or pituitary injury or disease |
| Cortisol | ACTH stimulation | A lack of response to prolonged ACTH infusion → adrenal insufficiency; response to ACTH → hypothalamic or pituitary diseases |
ACTH, adrenocorticotropic hormone; FSH, follicle-stimulating hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone.
Table 11-4 Endocrine Suppression Tests
| HORMONE OVER-PRODUCTION | TEST | EXPLANATION |
| Growth hormone | Oral glucose tolerance | Lack of suppression → pituitary adenoma |
| Cortisol | Dexamethasone suppression test | |
| Aldosterone | Isotonic saline infusion | Lack of suppression → aldosterone tumor |
ACTH, adrenocorticotropic hormone.
RADIOGRAPHIC IMAGING STUDIES
 Radioactive iodine uptake. High uptake in thyroid lesions is seen in hyperthyroidism due to Graves’ disease, toxic nodular goiter, or functioning solitary adenoma. Low uptake is seen in Hashimoto’s thyroiditis, tumors replacing normal thyroid tissue, and in patients who are taking exogenous thyroid hormones.
Radioactive iodine uptake. High uptake in thyroid lesions is seen in hyperthyroidism due to Graves’ disease, toxic nodular goiter, or functioning solitary adenoma. Low uptake is seen in Hashimoto’s thyroiditis, tumors replacing normal thyroid tissue, and in patients who are taking exogenous thyroid hormones.



Clinicopathologic Correlations
PITUITARY DISEASES
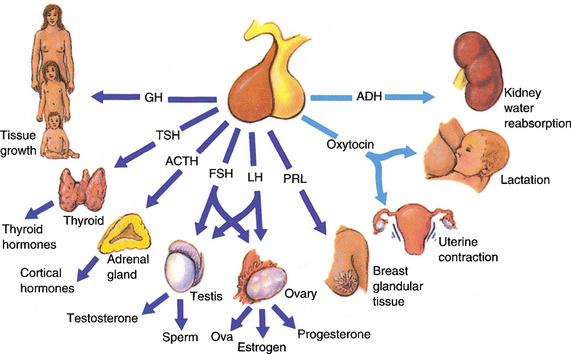
The pituitary gland is in central position to the hypothalamic–pituitary–peripheral endocrine organ system (Fig. 11-4). Accordingly, pituitary diseases resulting from the hyperproduction or hypoproduction of pituitary trophic hormones manifest with a hyperfunction or insufficiency of target organs or multiorgan endocrine failure.
Anterior pituitary hyperfunction is most often caused by tumors.
Prolactinoma. These tumors composed of lactotrophic cells account for approximately one third of all hormonally active pituitary adenomas. They occur eight times more frequently in women than in men. Hyperprolactinemia stimulates the production of milk, causing galactorrhea. It also interferes with the normally pulsatile secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), causing amenorrhea, anovulation, infertility, and loss of libido. In men it causes erectile dysfunction, loss of libido, infertility, and gynecomastia (Fig. 11-5).
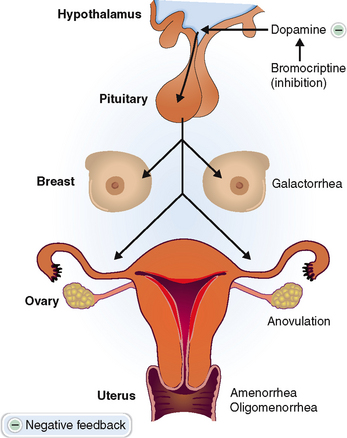
Tumors secreting prolactin may be classified as microadenomas or macroadenomas. Microadenomas do not affect the function of the surrounding normal pituitary, but the large macroadenomas may compress other cells and impede their function. The diagnosis is made by measuring prolactin in the blood and by demonstrating the tumor radiologically in the anterior lobe of the pituitary. Note, however, that hyperprolactinemia may be caused by a number of other conditions besides tumors (Table 11-5). In these conditions prolactin levels in the blood are usually less than 100 μg/L. A concentration of prolactin greater than 300 μg/L is virtually diagnostic of prolactinoma.
Table 11-5 Causes of Hyperprolactinemia
Somatotropic adenoma. These tumors secrete growth hormone. Childhood tumors found before the closure of epiphysis cause gigantism, and tumors of adulthood cause acromegaly. Gigantism is extremely rare, and acromegaly is somewhat more common. About 15% of all somatotropic adenomas also secrete prolactin and gonadotropic hormones.
Acromegaly. Acromegaly presents with acral enlargement due to thickening of the skin, excessive growth of subcutaneous tissue and bone, generalized organomegaly, and metabolic changes (Fig. 11-6). The most common findings include the following:
 Enlargement of hands and thickening of fingers, with palmar skin thickening. The patient typically complains that rings no longer fit or that gloves are tight. The patient also experiences excessive sweating.
Enlargement of hands and thickening of fingers, with palmar skin thickening. The patient typically complains that rings no longer fit or that gloves are tight. The patient also experiences excessive sweating.
 Enlargement of feet and toes and heel pad. The patient typically complains that shoes are too small.
Enlargement of feet and toes and heel pad. The patient typically complains that shoes are too small.





 Genital and reproductive problems. Loss of libido and erectile dysfunction are common in males, as is oligomenorrhea in women.
Genital and reproductive problems. Loss of libido and erectile dysfunction are common in males, as is oligomenorrhea in women.

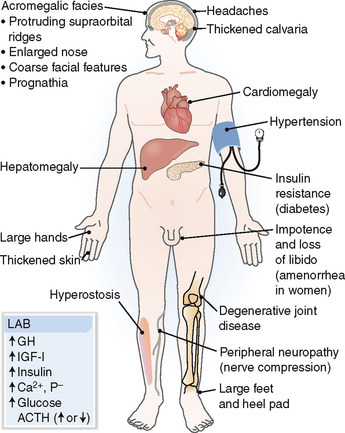
Figure 11-6 Acromegaly. ACTH, adrenocorticotropic hormone; GH, growth hormone; IGF-I, insulin-like growth factor I.
Hypopituitarism may be caused by many diseases affecting the anterior lobe of the pituitary, the pituitary stalk, or the hypothalamus.
A mnemonic for memorizing the causes of pituitary insufficiency is that all begin with a letter I (Table 11-6).
Table 11-6 Causes of Hypopituitarism
Data from Meszaros G: Endocrine and Reproductive Systems, Elsevier, 2006.
Diabetes insipidus is caused by a deficiency of antidiuretic hormone released from the posterior lobe of the pituitary.
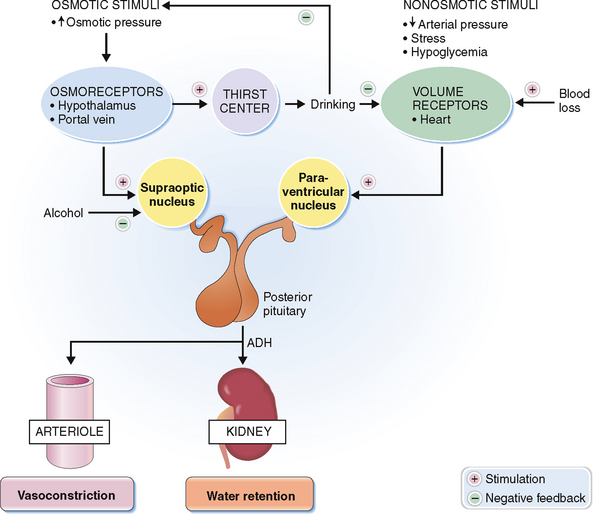
Antidiuretic hormone, also known as arginine vasopressin (AVP), is a nonapeptide synthesized by nerve cells and stored in the posterior lobe of the pituitary. It promotes the reabsorption of water in the collecting ducts of the kidneys (antidiuretic effect) and is a potent vasoconstrictor (vasopressin effect). It is released normally in response to increased osmolality of the plasma, which stimulates the osmoreceptors in the anterior hypothalamus (Fig. 11-7). Blood volume loss is yet another mechanism that leads to a release of ADH. This mechanism is initiated by low-pressure baroreceptors in the heart. Arterial hypotension, hypercapnia, hypoxia, pain, and nausea may all increase ADH release, causing vasoconstriction and antidiuresis.
Diabetes insipidus is a condition characterized by polyuria—excessive water loss through the kidneys. Two forms of diabetes insipidus are recognized (Fig. 11-8):
 Central diabetes insipidus. The inability to concentrate urine is related to a deficiency of ADH. It is a rare disease usually caused by destruction of hypothalamic nuclei, pituitary stalk, or neurohypophysis by tumors, trauma, or surgery (e.g., hypophysectomy for anterior pituitary tumors, brain tumors).
Central diabetes insipidus. The inability to concentrate urine is related to a deficiency of ADH. It is a rare disease usually caused by destruction of hypothalamic nuclei, pituitary stalk, or neurohypophysis by tumors, trauma, or surgery (e.g., hypophysectomy for anterior pituitary tumors, brain tumors).

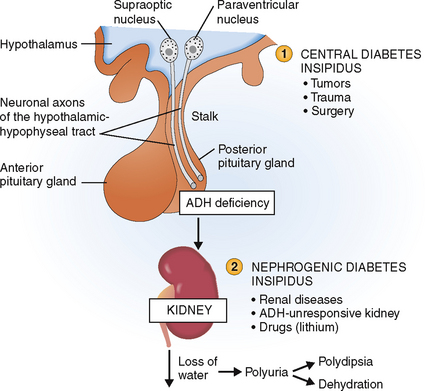
Figure 11-8 Diabetes insipidus. It may be central (1) or nephrogenic (2). ADH, antidiuretic hormone.
 Diabetic polyuria is characterized by hyperosmotic urine (>300 mOsm/kg). Hyperglycemia and glucosuria are readily demonstrable.
Diabetic polyuria is characterized by hyperosmotic urine (>300 mOsm/kg). Hyperglycemia and glucosuria are readily demonstrable.

The diagnosis of diabetes insipidus may be confirmed by the water deprivation test and/or injection of an analogue of ADH (desmopressin test). In patients with central diabetes insipidus water deprivation increases serum osmolality and raises the concentration of sodium, but does not increase the osmolality of the urine. Desmopressin injection increases urine osmolality over 50% from the baseline. In psychogenic polydipsia, water deprivation does not change serum osmolality and does not raise the serum sodium concentration, but the frequency of urination and the volume of urine increase, indicating that the kidney can concentrate urine. Desmopressin injection has no effect or only mildly increases serum osmolality (20–40% over the baseline).
Syndrome of inappropriate antidiuretic hormone secretion is characterized by water retention.
 Aberrant synthesis of ADH in malignant tumors (paraneoplastic syndrome)
Aberrant synthesis of ADH in malignant tumors (paraneoplastic syndrome)
 Synthesis of ADH by non-neoplastic lung tissue in chronic lung disorders
Synthesis of ADH by non-neoplastic lung tissue in chronic lung disorders
 Excessive release of ADH from the hypothalamus in brain disorders
Excessive release of ADH from the hypothalamus in brain disorders
The main causes of SIADH are listed in Table 11-7.
Table 11-7 Important Causes of Syndrome of Inappropriate ADH Secretion (SIADH)
ADH, antidiuretic hormone.
THYROID DISEASES
 Follicular epithelial cells secreting tyrosine-derived thyroid hormones and thyroglobulin
Follicular epithelial cells secreting tyrosine-derived thyroid hormones and thyroglobulin
 C cells secreting calcitonin, a polypeptide hormone involved in the metabolism of calcium
C cells secreting calcitonin, a polypeptide hormone involved in the metabolism of calcium
The secretion of thyroid hormones is under the control of the pituitary thyroid-stimulating hormone (TSH, or thyrotropin), which in turn is controlled by the hypothalamus secreting the thyrotropin-releasing hormone (TRH) (Fig. 11-9). Thyroid hormones provide a negative feedback inhibition to the hypothalamus and pituitary, inhibiting the secretion of TRH and TSH.
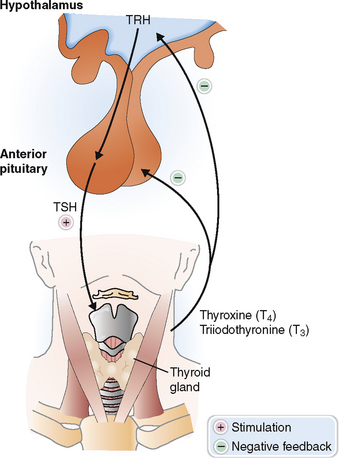
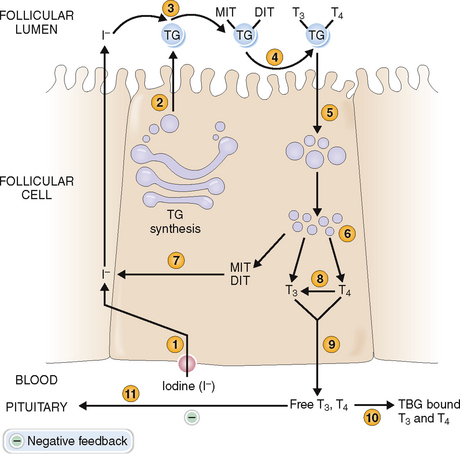
Follicular cells of the thyroid take up iodine and synthesize thyroid hormones from tyrosine.
Follicular epithelial cells synthesize thyroglobulin and secrete it into the lumen of the follicles as colloid (Fig. 11-10). The synthesis of thyroid hormones requires iodine, which is taken up by the follicular cells from the interstitial fluid (“iodine trapping”). Iodine is transported to the colloidal surface by a transport protein and “organified,” or attached to thyrosine to form monoiodotyrosine and diiodotyrosine. Coupling of these iodinated tyrosine derivatives leads to the formation of triiodotyronine (T3) and tetraiodothyronine (thyroxine, T4). Thyroglobulin is then endocytosed into the cytoplasm of follicular cells and cleaved by proteolysis, ultimately resulting in the formation of free T4. This process also leads to removal of iodine from monoiodothyronene and diiodothyronine molecules. T4 is converted in part to T3 by 5′-monodeiodinase, and both T3 and T4 are secreted into the circulation. Thyroxine predominates and accounts for the bulk of thyroid hormone secretion. Thus, only 15% of the total T3 is secreted in that form from the thyroid, whereas 85% of T3 is produced from circulating T4 in the periphery. Thyroxine can also be converted into a metabolically inactive form, reverse T3 (rT3). Serum levels of rT3 are typically elevated in acute nonthyroidal disease, reflecting a decreased concentration of T3.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


