Chapter 18 The Endocrine System
1 Which organs belong to the endocrine system?
The endocrine system consists of cells that secrete hormones and includes:







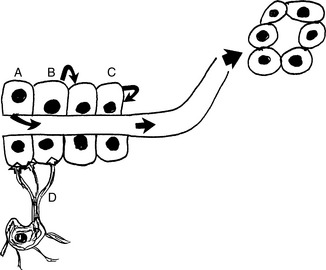



4 What are the differences between peptide and amine hormones versus steroid hormones?
In general, peptide and amine hormones are stored inside the cells of their origin in the form of dense neuroendocrine granules that can be seen by electron microscopy. These hormones act on receptors on the plasma membrane of target cells, which in turn activate a signaling system in the cytoplasm. The impulse is transmitted from the plasma membrane to the DNA by secondary messengers. In contrast to these hormones, steroids are not stored in granules but are synthesized on external stimulation by trophic hormones (e.g., adrenocorticotropic hormone [ACTH] stimulates the synthesis of glucocorticoids). Steroid hormones have receptors in the nucleus of their target cells, and their action on the nuclear DNA does not require activation of a cytoplasmic messenger system. See Fig. 18-2.



PITUITARY
10 What are the main cell types of the pituitary?









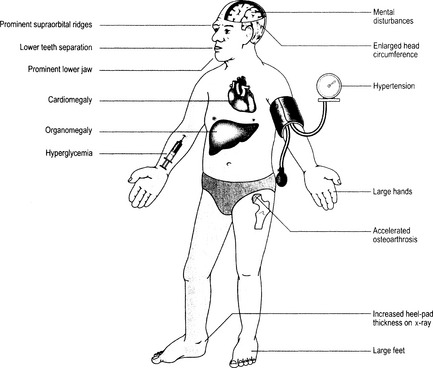
19 What are the features of acromegaly?
The most prominent findings are the marked enlargement of hands and feet, nose, and jaws (“acra,” i.e., protruding terminal parts). These externally visible changes are accompanied by internal organomegaly and metabolic disorders (diabetes, hypertension, etc.). The most common findings are illustrated in Fig. 18-3.
23 What is the anatomic substrate of hypopituitarism?
Clinical signs of hypopituitarism appear after a loss of 75% or more of the anterior pituitary.










THYROID
32 How is thyroxine formed?
37 What are the clinical features of hyperthyroidism?
 Cardiovascular system: Tachycardia, increased cardiac output, increased incidence of atrial fibrillation, and palpitations
Cardiovascular system: Tachycardia, increased cardiac output, increased incidence of atrial fibrillation, and palpitations




40 What are the possible causes of hypothyroidism?
Hypothyroidism may be related to three sets of causes:



For an extended list of possible causes of hypothyroidism, use the mnemonic add iodine:
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


