The stumbling way in which even the ablest of the scientists in every generation have had to fight through thickets of erroneous observations, misleading generalizations, inadequate formulations, and unconscious prejudice is rarely appreciated by those who obtain their scientific knowledge from textbooks.
–James Bryant Conant. From Science and Common Sense.
APPROACHES TO DRUG DEVELOPMENT
Nature of Drug Development
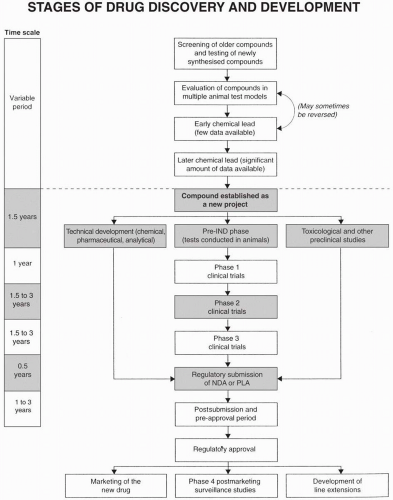
Drug development is the second major function of research and development groups. Drug discovery is the first function and marketing support, the third. Development is defined differently in different situations. It may specifically refer to technical aspects of chemical scaleup, analytical analysis of the active drug substance in a powder of the formulated product or in biological solutions or tissues, and pharmaceutical development of a dosage form and formulation. Alternatively, or in addition, the term development may refer to the entire process of taking a newly identified candidate compound for testing in humans through to regulatory approval for marketing (Fig. 10.1). This broad definition is used throughout this book. Many aspects of drug development are described in other chapters (e.g., technical development, clinical trials, and matrix concepts). A few additional points are presented below.
The importance of adhering to high regulatory and ethical standards in the conduct, analysis, and interpretation of all preclinical experiments and studies, as well as all clinical trials must be stressed. Poorly planned or conceived studies will often raise questions that will be difficult or impossible to explain adequately. Designing and conducting such studies is shortsighted, as it may take many months or years plus valuable resources to sort out the details and explain problems that did not have to arise in the first place. Yet, this is exactly what happens in many companies on a fairly frequent basis. In addition, the data these studies generate generally delay important regulatory submissions.
How Does a Company Decide Which Compounds to Develop as Drugs?
The simplest answer to this question, and also one that has a lot of truth, is that written or unwritten criteria are established (or should be established) for all research efforts directed at discovering a new drug. When a chemical compound is found that meets these criteria, then it is (usually) developed as a drug. The major criteria used to select a compound for development are listed in Table 10.1. The compound continues to be developed until events demonstrate that some of the criteria (primarily of safety and efficacy) cannot be met and that a medically and commercially viable drug is no longer likely or possible to achieve.
Because research programs in one therapeutic area may lead to a compound that has activity in another area, carefully developed criteria are not always present when a new direction in a drug’s development is debated. Considerations include determining how the compound fits into the company’s strategies and portfolio for current and future drug development. This includes evaluation of old product line-extensions, entrance into new product lines, perceived medical need, perceived commercial gains, plus the patent situation on the new compound.
Figure 10.1 Stages of drug discovery (above the dotted line) and development (below the dotted line). Various feedback loops exist in this diagram, especially during the initial stages of drug discovery. PLA, Product License Application.
In addition to the company’s basic strategies, there are issues about resources necessary to test and develop a new drug. Will it be relatively straightforward to evaluate the drug’s efficacy in humans, as with a diuretic or neuromuscular blocking agent, or will it be much more complex and expensive, as with an antipsychotic or anticancer agent? Is the path to regulatory approval different than that with an “average” small molecule drug (such as with a biotechnology created product)? In addition, some chemical classes of drugs with toxicity known to be greater than that of others in the same therapeutic class are not looked on favorably by regulatory authorities. In addition, each regulatory agency may require different types and degrees of proof of efficacy and safety. This will tend to make a drug’s development far more complex and costly for marketing it in one country than in another.
Table 10.1Research and development criteria to evaluate a new compound for potential development as a druga
Depending on the answers to these (and other) questions and criteria (Table 10.1), the company may decide to go all out, to go ahead with a limited program, to develop the drug only for certain countries, to license the drug to another company, or to take a different course of action.
Master Development Plan
Some companies prepare a comprehensive plan for a drug’s development as soon as the project and project team is formed. This usually occurs shortly after the compound is elevated to development status, which is about one to two years before the drug is evaluated in humans for totally new drugs and approximately six months to one year for drugs that have already been evaluated in patients in other countries. The same times generally apply for drugs licensed-in from other companies. This master plan is put together by the project leader or manager and becomes a “Bible” for many companies to follow. This document may be organized in any way a company desires or the format may be left to the author’s discretion.
One alternative is to prepare a detailed plan that only goes to the first milestone [e.g., submission of an Investigational New Drug Application (IND)], or includes a few milestones (e.g., regulatory agency meetings, plus Phase 1 and Phase 2a pilot studies). The author believes that this latter approach makes more sense than attempting to develop a detailed plan through to the submission of a marketing application at the outset of a program. In a few rare instances (e.g., some orphan drugs, or where a single study for a new indication is all that will be required) one would complete the entire master plan for development at the outset.
Part I
One approach is to present the overall development plan in three separate parts. The first part includes preclinical activities during the period from project initiation until early Phase 1 studies. This contains the background information nominating the compound for project status and discusses the pharmacology and toxicology programs. Alternatively, the compound may have been proposed for project status in a prior document that focused on the scientific, medical, and marketing rationales. A clinical feasibility section is included either in this part or as a separate document. Tables 111.1, 111.2, 111.3 in Guide to Clinical Trials (Spilker 1991) illustrate three examples of tables of contents of a clinical feasibility report.
Part II
Part two covers the period from initial regulatory submission (e.g., IND in the United States and a variety of applications elsewhere) to the end of Phase 2. This part contains the plan for metabolic studies, toxicology, regulatory affairs, chemical scaleup, supply sources, formulation, and clinical plan. The development of a product profile is considered, as are a variety of milestone dates that are used as targets within the company. Regulatory meetings such as a pre-IND meeting, end-of-Phase 1 and end-of-Phase 2, and pre-NDA meetings at the Food and Drug Administration (FDA) or other regulatory authority may also be tentatively noted on the calendar that is prepared. It is sometimes only relevant to create the initial part of the master plan as there may be so many questions to answer or issues to address that one is not able to write an entire plan at the outset of a project’s formation, but the entire plan would be completed as soon as it is possible to do so. The clinical development plan may include consideration of alternative paths, depending on the results of pilot studies. For example, if a Phase 2 study was to show the drug to be effective but also to have safety issues, plans may be developed to study lower doses or other indications. If the drug is found ineffective, but safe, plans may be proposed to study different patient populations with the same disease who may be more sensitive to the drug, or to initiate new pilot studies in different diseases.
A go-no-go decision point occurs in the development of many drugs toward the end of Phase 2. At that point, a sufficient amount of clinical efficacy and safety data are available to evaluate the drug, and the likelihood of its going to an NDA is assessed. If this probability is relatively high, it provides the impetus to commit company resources necessary for large-scale Phase 3 studies. There are certainly a few instances when such funds and an allout development effort are appropriate to initiate before proof of efficacy and knowledge of relative safety are obtained, but this decision should be made with full cognizance of the financial risks involved, as well as the awareness of which other drug development activities may be slowed or not pursued at all as a result of pursuing a drug with major resources and studies that has not actually reached its go-no-go decision point.
Part III
Part three covers the period from mid- or late-Phase 2 to registration filings or preferably to marketing. This part often contains a draft of the package insert, dosage forms to be developed, other aspects of technical development, details of manufacturing (including location, data processing, registration plans, and support functions), required clinical trials by country, specialized studies, and discussion of which expert opinions are required. Input on this section is required from all clinical, technical and other groups on the project development team. This part of the master plan will not be possible to write in detail until after the drug has entered or even completed Phase 2 studies.
Determining the Appropriate Magnitude of a Drug Development Plan: Lean versus Fat
A preclinical and clinical program of appropriate and well-designed studies may be planned to address all appropriate issues. A company may attempt to do this by developing a plan that emphasizes any desired approach varying from a “lean” one that eliminates all studies that are not absolutely essential to a “fat” approach that includes all possible considerations and studies. If the approach chosen is too “lean” and incomplete, it may not achieve regulatory approval in some or all of the target countries. A “fat” plan that contains many more studies than is regulatorily necessary would not only take extra years to complete, but would generate an excessive quantity of data that would require additional time for processing, analyzing, and interpretation.
How may a company know whether it has planned for studies that are an appropriate balance between these extremes? In many cases, the answer is difficult, but there are some principles to follow. First, determine if it would be reasonable to conduct any of the clinical trials after the drug is marketed. Second, determine what would be lost if each study on the list were deleted. Which studies are mandatory to conduct? Third, review the size of each study with a statistician. Could any be abbreviated? Fourth, discuss the clinical plan in detail with the FDA and/or other regulatory authorities. Additional steps might be (a) to evaluate the number and nature of studies conducted on recently approved drugs of the same type, (b) to discuss the issue with knowledgeable consultants, or (c) to develop alternative stepwise plans.
The master plan must distinguish between studies that are nice or desirable to conduct and those that are necessary to conduct. This principle applies to clinical, toxicological, metabolic, pharmacokinetic, and other categories of studies. The distinction between these two types of studies often changes during a drug’s development. Even though the master plan represents the blueprint of a drug’s development, the plans must frequently be reassessed and necessary changes made. This is discussed more extensively in the chapters in Section 5.
Establishing Criteria for a Drug’s Performance during Its Development
Compounds that have achieved the criteria established during the drug discovery period are advanced into development. Even if clear criteria have not been established preclinically during the discovery period, it is important to set criteria during development. This is primarily to know when the drug’s development should be terminated.
Criteria established at an early stage of development to judge each drug’s future performance should include medical, technical, and commercial considerations. If the drug falls below or otherwise fails to meet these standards, then its termination should be considered. This does not mean that a company would not develop one or more orphan drugs where there is little hope of ever repaying their development costs. However, it does mean that appropriate standards should be established for orphan drugs, just as for others. Thus, the company’s management would agree to accept lower minimal medical and/or commercial standards for some drugs. If medical and/or commercial criteria are established, a project’s termination would be expedited if the drug’s characteristics demonstrated that the minimally acceptable criteria could not be met.
The criteria used to determine whether a drug passes the go-no-go decision point in Phase 2 may be set to a particularly high or low standard. For example, the marketing group may believe they do not want to sell a drug unless it is sensational medically, or they may believe that it is desirable to have the drug available for sale, even if it is not as effective or safe as the medical staff and managers hope it will be. Whichever standards are chosen, they should be carefully scripted so that it is apparent to all if the profile obtained meets or does not meet these standards.
An important new drug with great medical value is of limited commercial benefit to a company if actual sales are small or modest, but this type of drug may provide other benefits to a company (e.g., enhanced reputation and improved access to physicians by sales representatives). The company’s overall reactions to drugs with limited commercial potential, as well as other drugs, depends to a large degree on their original expectations. These expectations relate in many ways to the criteria established for the drug’s development.
Only gold members can continue reading. Log In or Register to continue