sinuses, which result from bifurcations of the nutrient or medullary arteries, are the basic structural unit of the bone marrow (12). Within this histologic compartment, HSC and progenitor cells are exposed to the extracellular matrix that comprises the bone marrow microenvironment (Figure 24-1). The outer adventitial reticular cells (ARCs) add connective tissue elements and form the outer sinusoidal wall and synthesize collagen, laminin, fibronectin, and proteoglycans. All regulatory factors, adhesion molecules, and other proteins necessary for the regulation of hematopoiesis are contained within this matrix (1,13). Furthermore, the ARCs are phagocytic and can become lipocytes. As outlined before, the fat/hematopoietic ratio (“marrow cellularity”) is variable and a rough estimate can be calculated as cellularity = 100% — age (see below). Mitotically active cells are normally found around the supporting bone, typically paratrabecular and perivascular from where cells mature progressively into the medullary cavity. All newly formed mature hematopoietic cells are released into the bone marrow capillary-venous sinuses. Most cells pass through the sinus wall, but megakaryocytes reside adjacent to sinuses and extend pseudopodia directly into the vascular space (14,15). The capillary-venous sinuses coalesce into the venules and ultimately become veins that carry newly formed hematopoietic cells to the systemic circulation (12).
TABLE 24-1 GENERAL FEATURES OF THE BONE MARROW AND HEMATOPOIESIS | ||
|---|---|---|
|
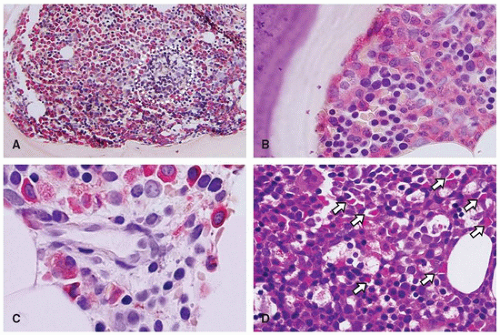 FIGURE 24-1 • Bone marrow microarchitecture. A: Bone marrow biopsy from a 1-day-old boy showing hematopoietic tissue that occupies approximately 90% of the marrow space. Only few regions of bone marrow fat are seen. The myeloid lineage is highlighted in red (Leder stain), and the perivascular region (circle) shows lack of myeloid cells. B: The paratrabecular region shows myeloid and erythroid precursors. C: Perivascular distribution of precursors in a bone marrow biopsy from an 18-year-old girl; note the delicate reticulum and extracellular matrix derived from ARCs. D: Highly cellular (>90%) bone marrow biopsy in a preterm girl shows numerous capillaries (arrows) interspersed between the hematopoietic cells and extracellular matrix. |
stimulatory activities of colony-stimulating factors (CSFs) and interleukins (ILs), whereas antagonistic effects are driven by inhibitory factors that include tumor necrosis factor (18,19). It is known that mature hematopoietic elements play a role in the regulation of lineage production and in maintaining steady-state hematopoiesis. In addition to the complicated molecular pathways, endocrine, paracrine, mesenchymal, and autonomic nervous system regulations have been implicated for homeostasis (18,19).
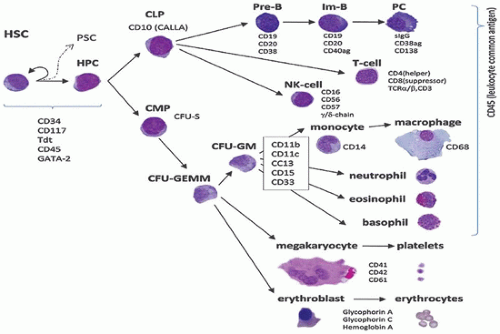 FIGURE 24-2 • Selected aspects of hematopoiesis. See text for details. CLP, committed lymphoid progenitor; CMP, committed myeloid progenitor (e.g., CFU-S: colony-forming unit—spleen); GEMM, granulo-erythro-megakaryo-monocytic; GM, granulo-monocytic (= myelomonocytic); HPC, hematopoietic progenitor committed; HSC, hematopoietic stem cell; Im-B, immature B-lymphocyte; PC, plasma cell; PSC, peripheral stem cell. |
and neutrophils comprise the largest maturation storage compartment of the bone marrow, which can be released into the peripheral blood in response to multiple host-mediated challenges.
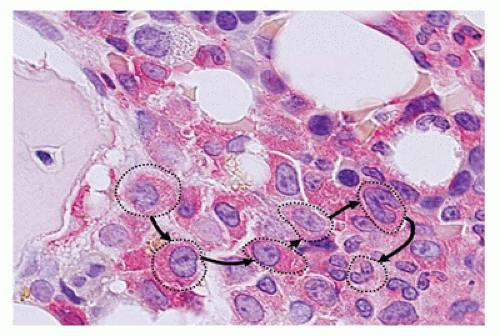 FIGURE 24-3 • Granulopoiesis. Immature granulocytic precursors (Leder positive) localize to the paratrabecular regions. Subsequent arbitrary stages are indicated (circles) and maturation progresses to, for example, band neutrophils. |
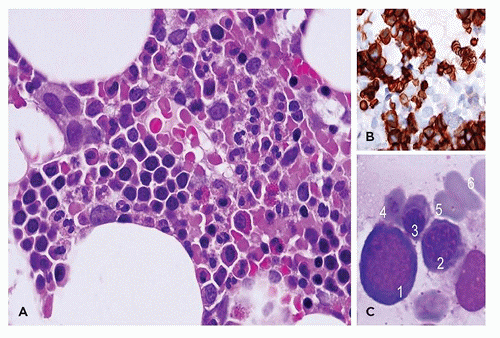 FIGURE 24-4 • A: Erythropoiesis occurs in small colonies (erythroblast islands) related to vascular structures. B: Glycophorin A; marker of erythroid differentiation. C: Subsequent stages of erythroid differentiation. |
and the acquisition of cytoplasmic granules characterize the stages of maturation, designated as monoblast, promonocyte, and mature monocyte. Although characteristically, monocytes have fewer and smaller granules than neutrophils, neither monoblasts nor promonocytes are generally recognizable in normal bone marrow. Monocytes circulate in the blood and subsequently migrate to solid tissues and become macrophages or various types of immune accessory cells. Due to this accessory role and evidence that these cells play an integrated, multifaceted role in humoral and cellular immunity beyond simple phagocytosis, the former designation mononuclear phagocyte system (37) has been replaced. Foucar and Foucar (38) proposed the alternative name mononuclear phagocyte and immunoregulatory effector (M-PIRE) system as a more accurate descriptor. The M-PIRE system includes monocytes, macrophages, multiple dendritic cells (e.g., Langerhans and dendritic reticulum cells), and their bone marrow precursors. Some evidence suggests a common cell of origin (39). Because the constituent cells show unique immunophenotypic and functional properties, the M-PIRE designation remains controversial. Regardless of the name, both macrophages (histiocytes) and dendritic cells are inconspicuous normal constituents of virtually all organ systems, and mature cells of monocyte-macrophage lineage also remain as a major constituent of the bone marrow microenvironment.
generally stable. A dramatic decline in erythroid elements parallels the drop in EPO levels that occurs after birth in full-term neonates (49). Erythropoiesis returns to normal steady-state levels following resolution of this so-called physiologic anemia of infancy. Likewise, dramatic age-related variations occur in the proportion of bone marrow lymphoid cells, with up to 40% lymphocytes in bone marrow specimens of very young children and infants (50). The proportion of lymphocytes decreases in bone marrow specimens, and B-cell production in general declines with age (51).
TABLE 24-2 NORMAL VALUES FOR BONE MARROW AND DIFFERENTIAL CELL COUNTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 24-3 HEMATOLOGIC PROFILE DURING THE FIRST MONTH OF LIFE AND IN YOUNG INFANTS | |
|---|---|
|
TABLE 24-4 INDICATIONS FOR BONE MARROW EXAMINATION IN CHILDREN | |
|---|---|
|
TABLE 24-5 SPECIALIZED TECHNIQUES IN BONE MARROW EXAMINATIONS | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
beyond the first 3 to 4 days of life (53). In healthy neonates, the relative hypoxia in utero is reversed at birth, so that a marked, transient, abrupt decline in erythropoiesis (so-called physiologic anemia of infancy) occurs. These physiologic changes are exaggerated in preterm infants (49).
TABLE 24-6 CONSTITUTIONAL HEMATOLOGIC DISORDERS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
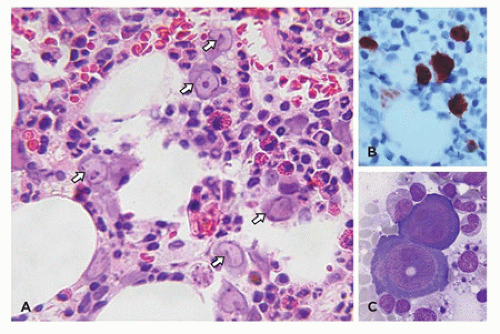 FIGURE 24-5 • Parvovirus. A,B: Intranuclear viral inclusions in a patient with parvo B19-induced red cell aplasia (arrows) and corresponding parvovirus immunohistochemistry. C: Morphology of intranuclear inclusion on smear (Wright-Giemsa stain). |
morphologic features of parvovirus infection are lacking. Another distinctive bone marrow finding, increased hematogones (74,75), may accompany any type of red cell aplasia in children, especially in very young patients.
TABLE 24-7 SELECTED CONSTITUTIONAL HEMATOLOGIC DISORDERS INVOLVING GRANULOCYTES | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


