The Arrhythmias
KEY CONCEPTS
![]() The use of antiarrhythmic drugs (AADs) in the United States has declined because of major trials that show increased mortality with their use in several clinical situations, the realization of proarrhythmia as a significant side effect, and the advancing technology of nonpharmacologic therapies such as ablation and the implantable cardioverter-defibrillator (ICD).
The use of antiarrhythmic drugs (AADs) in the United States has declined because of major trials that show increased mortality with their use in several clinical situations, the realization of proarrhythmia as a significant side effect, and the advancing technology of nonpharmacologic therapies such as ablation and the implantable cardioverter-defibrillator (ICD).
![]() AADs frequently cause side effects and are complex in their pharmacokinetic characteristics. Close monitoring is required of all of these drugs to assess for adverse effects as well as potential drug interactions.
AADs frequently cause side effects and are complex in their pharmacokinetic characteristics. Close monitoring is required of all of these drugs to assess for adverse effects as well as potential drug interactions.
![]() The most commonly prescribed AAD is now amiodarone. This drug is effective in terminating and preventing a wide variety of symptomatic supraventricular and ventricular arrhythmias. However, because this AAD is plagued by frequent side effects, it requires close monitoring. The most concerning toxicity is pulmonary fibrosis; side effect profiles of the IV (acute, short-term) and oral (chronic, long-term) forms of amiodarone differ substantially.
The most commonly prescribed AAD is now amiodarone. This drug is effective in terminating and preventing a wide variety of symptomatic supraventricular and ventricular arrhythmias. However, because this AAD is plagued by frequent side effects, it requires close monitoring. The most concerning toxicity is pulmonary fibrosis; side effect profiles of the IV (acute, short-term) and oral (chronic, long-term) forms of amiodarone differ substantially.
![]() In patients with atrial fibrillation (AF), therapy is traditionally aimed at controlling ventricular rate (digoxin, nondihydropyridine calcium channel blockers, β-blockers), preventing thromboembolic complications (warfarin, aspirin), and restoring and maintaining sinus rhythm (AADs, direct current cardioversion). Studies show there is no need to aggressively pursue strategies to maintain sinus rhythm (i.e., long-term AAD therapy); rate control alone (leaving the patient in AF) is often sufficient in patients who can tolerate it. Nonetheless, chronic AAD therapy may still be needed in patients who continue to have symptoms despite adequate ventricular rate control.
In patients with atrial fibrillation (AF), therapy is traditionally aimed at controlling ventricular rate (digoxin, nondihydropyridine calcium channel blockers, β-blockers), preventing thromboembolic complications (warfarin, aspirin), and restoring and maintaining sinus rhythm (AADs, direct current cardioversion). Studies show there is no need to aggressively pursue strategies to maintain sinus rhythm (i.e., long-term AAD therapy); rate control alone (leaving the patient in AF) is often sufficient in patients who can tolerate it. Nonetheless, chronic AAD therapy may still be needed in patients who continue to have symptoms despite adequate ventricular rate control.
![]() Paroxysmal supraventricular tachycardia is usually a result of reentry in or proximal to the atrioventricular (AV) node or AV reentry incorporating an extranodal pathway; common tachycardias can be terminated acutely with AV nodal blocking drugs such as adenosine, and recurrences can be prevented by ablation with radio-frequency current.
Paroxysmal supraventricular tachycardia is usually a result of reentry in or proximal to the atrioventricular (AV) node or AV reentry incorporating an extranodal pathway; common tachycardias can be terminated acutely with AV nodal blocking drugs such as adenosine, and recurrences can be prevented by ablation with radio-frequency current.
![]() Patients with Wolff-Parkinson-White (WPW) syndrome may have several different tachycardias that are acutely treated by different strategies: orthodromic reentry (adenosine), antidromic reentry (adenosine or procainamide), and AF (procainamide or amiodarone). AV nodal blocking drugs are contraindicated in patients with WPW syndrome and AF.
Patients with Wolff-Parkinson-White (WPW) syndrome may have several different tachycardias that are acutely treated by different strategies: orthodromic reentry (adenosine), antidromic reentry (adenosine or procainamide), and AF (procainamide or amiodarone). AV nodal blocking drugs are contraindicated in patients with WPW syndrome and AF.
![]() Because of the results of the Cardiac Arrhythmia Suppression Trial and other trials, AADs (with the exception of β-blockers) should not be routinely used in patients with prior myocardial infarction (MI) or left ventricular (LV) dysfunction and minor ventricular rhythm disturbances (e.g., premature ventricular complexes).
Because of the results of the Cardiac Arrhythmia Suppression Trial and other trials, AADs (with the exception of β-blockers) should not be routinely used in patients with prior myocardial infarction (MI) or left ventricular (LV) dysfunction and minor ventricular rhythm disturbances (e.g., premature ventricular complexes).
![]() Patients with hemodynamically significant ventricular tachycardia (VT) or ventricular fibrillation not associated with an acute MI who are successfully resuscitated (electrical cardioversion, vasopressors, amiodarone) are at high risk for sudden cardiac death (SCD) and should receive an ICD (“secondary prevention”).
Patients with hemodynamically significant ventricular tachycardia (VT) or ventricular fibrillation not associated with an acute MI who are successfully resuscitated (electrical cardioversion, vasopressors, amiodarone) are at high risk for sudden cardiac death (SCD) and should receive an ICD (“secondary prevention”).
![]() Implantation of an ICD should be considered for the primary prevention of SCD in certain high-risk patient populations. High-risk patients include those with a history of MI and LV dysfunction (regardless of whether they have inducible sustained ventricular arrhythmias), as well as those with New York Heart Association class II or III heart failure as a result of either ischemic or nonischemic causes.
Implantation of an ICD should be considered for the primary prevention of SCD in certain high-risk patient populations. High-risk patients include those with a history of MI and LV dysfunction (regardless of whether they have inducible sustained ventricular arrhythmias), as well as those with New York Heart Association class II or III heart failure as a result of either ischemic or nonischemic causes.
![]() Life-threatening ventricular proarrhythmia generally takes two forms: sinusoidal or incessant monomorphic VT (class Ic AADs) and torsade de pointes (class Ia or III AADs and many other noncardiac drugs).
Life-threatening ventricular proarrhythmia generally takes two forms: sinusoidal or incessant monomorphic VT (class Ic AADs) and torsade de pointes (class Ia or III AADs and many other noncardiac drugs).
The heart has two basic properties, namely, an electrical property and a mechanical property. The synchronous interaction between these two properties is complex, precise, and relatively enduring. The study of the electrical properties of the heart has grown at a steady rate, interrupted by periodic salvos of scientific breakthroughs. Einthoven’s pioneering work allowed graphic electrical tracings of cardiac rhythm and probably represents the first of these breakthroughs. This discovery of the surface electrocardiogram (ECG) has remained the cornerstone of diagnostic tools for cardiac rhythm disturbances. Since then, intracardiac recordings and programmed cardiac stimulation have advanced our understanding of arrhythmias, and microelectrode, voltage clamping, and patch clamping techniques have allowed considerable insight into the electrophysiologic actions and mechanisms of antiarrhythmic drugs (AADs). Certainly, the new era of molecular biology and mapping of the human genome promises even greater insights into mechanisms (and potential therapies) of arrhythmias. Noteworthy in this regard is the discovery of genetic abnormalities in the ion channels that control electrical repolarization (heritable long QT syndrome) or depolarization (Brugada syndrome).
The clinical use of drug therapy started with the use of digitalis and then quinidine. A surge of new agents followed somewhat later in the 1980s. A theme of drug discovery during this decade was initially to find orally absorbed lidocaine congeners (such as mexiletine and tocainide); later, the emphasis was on drugs with extremely potent effects on conduction (i.e., flecainide-like agents). The most recent focus of investigational AADs is the potassium channel blockers, with dronedarone being the most recently approved AAD in the United States in nearly a decade. Previously, there was some expectation that advances in AAD discovery would lead to a highly effective and nontoxic agent that would be effective for a majority of patients (i.e., the so-called magic bullet). Instead, significant problems with drug toxicity and proarrhythmia have resulted in a decline in the overall volume of AAD usage in the United States since 1989.
![]() The other phenomenon that has significantly contributed to the decline in AAD utilization is the development of extremely effective nonpharmacologic therapies. Technical advances have made it possible to permanently interrupt reentry circuits with radiofrequency ablation, which renders long-term AAD use unnecessary in certain arrhythmias. Furthermore, the impressive survival data associated with the use of implantable cardioverter-defibrillators (ICDs) for the primary and secondary prevention of sudden cardiac death (SCD) have led most clinicians to choose “device” therapy as the first-line treatment for patients who are at high risk for life-threatening ventricular arrhythmias. Both of these nonpharmacologic therapies have become increasingly popular for the management of arrhythmias so that the potential proarrhythmic effects and organ toxicities associated with AADs can be avoided.
The other phenomenon that has significantly contributed to the decline in AAD utilization is the development of extremely effective nonpharmacologic therapies. Technical advances have made it possible to permanently interrupt reentry circuits with radiofrequency ablation, which renders long-term AAD use unnecessary in certain arrhythmias. Furthermore, the impressive survival data associated with the use of implantable cardioverter-defibrillators (ICDs) for the primary and secondary prevention of sudden cardiac death (SCD) have led most clinicians to choose “device” therapy as the first-line treatment for patients who are at high risk for life-threatening ventricular arrhythmias. Both of these nonpharmacologic therapies have become increasingly popular for the management of arrhythmias so that the potential proarrhythmic effects and organ toxicities associated with AADs can be avoided.
This chapter reviews the principles involved in both normal and abnormal cardiac conduction and addresses the pathophysiology and treatment of the more commonly encountered arrhythmias. Certainly, many volumes of complete text could be (and have been) devoted to basic and clinical electrophysiology. Consequently, this chapter briefly addresses those principles necessary for clinicians.
ARRHYTHMOGENESIS
Normal Conduction
Electrical activity is initiated by the sinoatrial (SA) node and moves through cardiac tissue by a tree-like conduction network. The SA node initiates cardiac rhythm under normal circumstances because this tissue possesses the highest degree of automaticity or rate of spontaneous impulse generation. The degree of automaticity of the SA node is largely influenced by the autonomic nervous system in that both cholinergic and sympathetic innervations control the sinus rate. Most tissues within the conduction system also possess varying degrees of inherent automatic properties. However, the rates of spontaneous impulse generation of these tissues are less than that of the SA node. Thus, these latent automatic pacemakers are continuously overdriven by impulses arising from the SA node (primary pacemaker) and do not become clinically apparent.
From the SA node, electrical activity moves in a wave front through an atrial specialized conducting system and eventually gains entrance to the ventricle via the atrioventricular (AV) node and a large bundle of conducting tissue referred to as the bundle of His. Aside from this AV node–bundle of His pathway, a fibrous AV ring that will not permit electrical stimulation separates the atria and ventricles. The conducting tissues bridging the atria and ventricles are referred to as the junctional areas. Again, this area of tissue (junction) is largely influenced by autonomic input and possesses a relatively high degree of inherent automaticity (about 40 beats/min, less than that of the SA node). From the bundle of His, the cardiac conduction system bifurcates into several (usually three) bundle branches: one right bundle and two left bundles. These bundle branches further arborize into a conduction network referred to as the Purkinje system. The conduction system as a whole innervates the mechanical myocardium and serves to initiate excitation–contraction coupling and the contractile process. After a cell or group of cells within the heart is electrically stimulated, a brief period of time follows in which those cells cannot again be excited. This time period is referred to as the refractory period. As the electrical wave front moves down the conduction system, the impulse eventually encounters tissue refractory to stimulation (recently excited) and subsequently dies out. The SA node subsequently recovers, fires spontaneously, and begins the process again.
Prior to cellular excitation, an electrical gradient exists between the inside and the outside of the cell membrane. At this time, the cell is polarized. In atrial and ventricular conducting tissues, the intracellular space is approximately 80 to 90 mV negative with respect to the extracellular environment. The electrical gradient just prior to excitation is referred to as the resting membrane potential (RMP) and is the result of differences in ion concentrations between the inside and the outside of the cell. At RMP, the cell is polarized primarily by the action of active membrane ion pumps, the most notable of these being the sodium–potassium pump. For example, this specific pump (in addition to other systems) attempts to maintain the intracellular sodium concentration at 5 to 15 mEq/L and the extracellular sodium concentration at 135 to 142 mEq/L, and the intracellular potassium concentration at 135 to 140 mEq/L and the extracellular potassium concentration at 3 to 5 mEq/L. The RMP can be calculated by using the Nernst equation:
![]()
Electrical stimulation (or depolarization) of the cell will result in changes in membrane potential over time or a characteristic action potential curve (Fig. 8-1). The action potential curve results from the transmembrane movement of specific ions and is divided into different phases. Phase 0 or initial, rapid depolarization of atrial and ventricular tissues is caused by an abrupt increase in the permeability of the membrane to sodium influx. This rapid depolarization more than equilibrates (overshoots) the electrical potential, resulting in a brief initial repolarization or phase 1. Phase 1 (initial repolarization) is caused by a transient and active potassium efflux (i.e., the IKto current). Calcium begins to move into the intracellular space at about –60 mV (during phase 0), causing a slower depolarization. Calcium influx continues throughout phase 2 of the action potential (plateau phase) and is balanced to some degree by potassium efflux. Calcium entrance (only through L channels in myocardial tissue) distinguishes cardiac conducting cells from nerve tissue and provides the critical ionic link to excitation–contraction coupling and the mechanical properties of the heart as a pump. The membrane remains permeable to potassium efflux during phase 3, resulting in cellular repolarization. Phase 4 of the action potential is the gradual depolarization of the cell and is related to a constant sodium leak into the intracellular space balanced by a decreasing (over time) efflux of potassium. The slope of phase 4 depolarization determines, in large part, the automatic properties of the cell. As the cell is slowly depolarized during phase 4, an abrupt increase in sodium permeability occurs, allowing the rapid cellular depolarization of phase 0. The juncture of phase 4 and phase 0 where rapid sodium influx is initiated is referred to as the threshold potential of the cell. The level of threshold potential also regulates the degree of cellular automaticity.

FIGURE 8-1 Purkinje fiber action potential showing specific ion flux responsible for the change in membrane potential. Ions outside of the line (e.g., sodium) move from the extracellular space to the intracellular space and ions on the inside of the line (e.g., potassium) move from the inside of the cell to the outside.
Not all cells in the cardiac conduction system rely on sodium influx for initial depolarization. Some tissues depolarize in response to a slower inward ionic current caused by calcium influx. These “calcium-dependent” tissues are found primarily in the SA and AV nodes (both L and T channels) and possess distinct conduction properties in comparison to “sodium-dependent” fibers. Calcium-dependent cells generally have a less negative RMP (–40 to –60 mV) and a slower conduction velocity. Furthermore, in calcium-dependent tissues, recovery of excitability outlasts full repolarization, whereas in sodium-dependent tissues, recovery is prompt after repolarization. These two types of electrical fibers also differ dramatically in how drugs modify their conduction properties.
Ion conductance across the lipid bilayer of the cell membrane occurs via the formation of membrane pores or “channels” (Fig. 8-2). Selective ion channels probably form in response to specific electrical potential differences between the inside and the outside of the cell (voltage dependence). The membrane itself is composed of both organized and disorganized lipids and phospholipids in a dynamic sol-gel matrix. During ion flux and electrical excitation, changes in this sol-gel equilibrium occur and permit the formation of activated ion channels. Besides channel formation and membrane composition, intrachannel proteins or phospholipids, referred to as gates, also regulate the transmembrane movement of ions. These gates are thought to be positioned strategically within the channel to modulate ion flow (Fig. 8-2). Each ion channel conceptually has two types of gates: an activation gate and an inactivation gate. The activation gate opens during depolarization to allow the ion current to enter or exit from the cell, and the inactivation gate later closes to stop ion movement. When the cell is in a rested state, the activation gates are closed and the inactivation gates are open. The activation gates then open to allow ion movement through the channel, and the inactivation gates later close to stop ion conductance. Thus, the cell cycles between three states: resting, activated or open, and inactivated or closed. Activation of SA and AV nodal tissue is dependent on a slow depolarizing current through calcium channels and gates, whereas the activation of atrial and ventricular tissues is dependent on a rapid depolarizing current through sodium channels and gates.
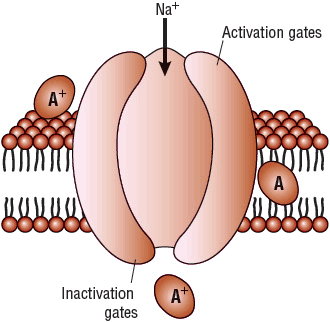
FIGURE 8-2 Lipid bilayer, sodium channel, and possible sites of action of the class I AADs (A). Class I AADs may theoretically inhibit sodium influx at an extracellular, intramembrane, or intracellular receptor site. However, all approved agents appear to block sodium conductance at a single receptor site by gaining entrance to the interior of the channel from an intracellular route. Active ionized drugs block the channel predominantly during the activated or inactivated state and bind and unbind with specific time constants (described as fast on–off, slow on–off, and intermediate). (AADs, antiarrhythmic drugs.)
Abnormal Conduction
The mechanisms of tachyarrhythmias have been classically divided into two general categories: those resulting from an abnormality in impulse generation or “automatic” tachycardias and those resulting from an abnormality in impulse conduction or “reentrant” tachycardias.
Automatic tachycardias depend on spontaneous impulse generation in latent pacemakers and may be a result of several different mechanisms. Drugs, such as digoxin or catecholamines, and conditions, such as hypoxia, electrolyte abnormalities (e.g., hypokalemia), and fiber stretch (cardiac dilation), may lead to an increased slope of phase 4 depolarization in cardiac tissues other than the SA node. These factors that experimentally lead to abnormal automaticity are also known to be arrhythmogenic in clinical situations. The increased slope of phase 4 causes heightened automaticity of these tissues and competition with the SA node for dominance of cardiac rhythm. If the rate of spontaneous impulse generation of the abnormally automatic tissue exceeds that of the SA node, then an automatic tachycardia may result. Automatic tachycardias have the following characteristics: (a) the onset of the tachycardia is unrelated to an initiating event such as a premature beat; (b) the initiating beat is usually identical to subsequent beats of the tachycardia; (c) the tachycardia cannot be initiated by programmed cardiac stimulation; and (d) the onset of the tachycardia is usually preceded by a gradual acceleration in rate and termination is usually preceded by a gradual deceleration in rate. Clinical tachycardias resulting from the classic forms of enhanced automaticity already described are not as common as once thought. Examples are sinus tachycardia and junctional tachycardia.
Triggered automaticity is also a possible mechanism for abnormal impulse generation. Briefly, triggered automaticity refers to transient membrane depolarizations that occur during repolarization (early afterdepolarizations [EADs]) or after repolarization (late afterdepolarizations [LADs]) but prior to phase 4 of the action potential. Afterdepolarizations may be related to abnormal calcium and sodium influx during or just after full cellular repolarization. Experimentally, EADs may be precipitated by hypokalemia, class Ia AADs, or slow stimulation rates—any factor that blocks the ion channels (e.g., potassium) responsible for cellular repolarization. EADs provoked by drugs that block potassium conductance and delay repolarization are the underlying cause of torsade de pointes (TdP). LADs may be precipitated by digoxin or catecholamines and suppressed by calcium channel blockers (CCBs), and have been suggested as the mechanism for multifocal atrial tachycardia, digoxin-induced tachycardias, and exercise-provoked ventricular tachycardia (VT). Triggered automatic rhythms possess some of the characteristics of automatic tachycardias and some of the characteristics of reentrant tachycardias (description follows).
Reentry is a concept that involves indefinite propagation of the impulse and continued activation of previously refractory tissue. There are three conduction requirements for the formation of a viable reentrant focus: 1) two pathways for impulse conduction, 2) an area of unidirectional block (prolonged refractoriness) in one of these pathways, and 3) slow conduction in the other pathway (Fig. 8-3). Usually, a critically timed premature beat initiates reentry. This premature impulse enters both conduction pathways but encounters refractory tissue in one of the pathways at the area of unidirectional block. The impulse dies out because the tissue is still refractory from the previous (sinus) impulse. Although it fails to propagate in one pathway, the impulse may still proceed in a forward direction (antegrade) through the other pathway because of this pathway’s relatively shorter refractory period. The impulse may then proceed through a loop of tissue and “reenter” the area of unidirectional block in a backward direction (retrograde). Because the antegrade pathway has slow conduction characteristics, the area of unidirectional block has time to recover its excitability. The impulse can proceed in a retrograde fashion through this (previously refractory) tissue and continue around the loop of tissue in a circular fashion. Thus, the key to the formation of a reentrant focus is crucial conduction discrepancies in the electrophysiologic characteristics of the two pathways. The reentrant focus may excite surrounding tissue at a rate greater than that of the SA node, leading to formation of a clinical tachycardia. The above model is anatomically determined in that there is only one pathway for impulse conduction with a fixed circuit length. Another model of reentry, referred to as a functional reentrant loop or leading circle model, may also occur (Fig. 8-4).1 In a functional reentrant focus, the length of the circuit may vary depending on the conduction velocity and recovery characteristics of the impulse. The area in the middle of the loop is continually kept refractory by the inwardly moving impulse. The length of the circuit is not fixed but is the smallest circle possible, such that the leading edge of the wave front is continuously exciting tissue just as it recovers, that is, the head of the impulse nearly catches its tail. It differs from the anatomic model in that the leading edge of the impulse is not preceded by an excitable gap of tissue, and it does not have an obstacle in the middle or a fixed anatomic circuit. Clinically, many reentrant foci probably have both anatomic and functional characteristics. In the figure 8 model, a zone of unidirectional block is present, allowing for two impulse loops that join and reenter the area of block in a retrograde fashion to form a pretzel-shaped reentrant circuit. This model combines functional characteristics with an excitable gap. All of these theoretical models require a critical balance of refractoriness and conduction velocity within the circuit and as such have helped to explain the effects of drugs on terminating, modifying, and causing cardiac rhythm disturbances.
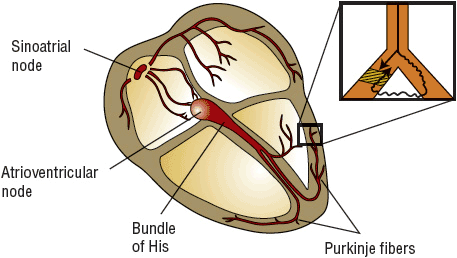
FIGURE 8-3 Conduction system of the heart. The magnified portion shows a bifurcation of a Purkinje fiber traditionally explained as the etiology of reentrant VT. A premature impulse travels to the fiber, damaged by heart disease or ischemia. It encounters a zone of prolonged refractoriness (area of unidirectional block; cross-hatched area) but fails to propagate because it remains refractory to stimulation from the previous impulse. However, the impulse may slowly travel (squiggly line) through the other portion of the Purkinje twig and will “reenter” the cross-hatched area if the refractory period is concluded and it is now excitable. Thus, the premature impulse never meets refractory tissue; circus movement ensues. If this site stimulates the surrounding ventricle repetitively, clinical reentrant VT results. (VT, ventricular tachycardia.)
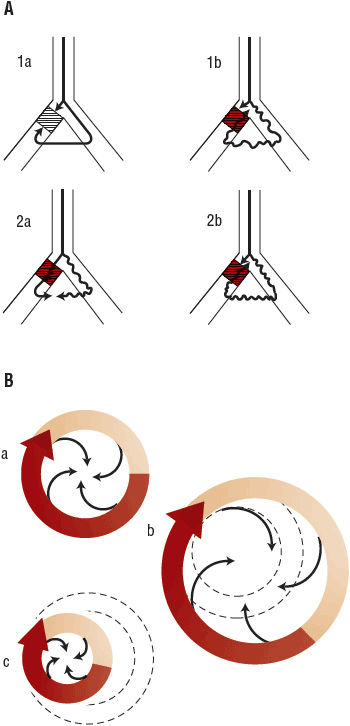
FIGURE 8-4 A. Possible mechanism of proarrhythmia in the anatomic model of reentry. 1a. Nonviable reentrant loop due to bidirectional block (shaded area). 1b. Instance where a drug slows conduction velocity without significantly prolonging the refractory period. The impulse is now able to reenter the area of unidirectional block (shaded area) because slowed conduction through the contralateral limb allows recovery of the block. A new reentrant tachycardia may result. 2a. Nonviable reentrant loop due to a lack of a unidirectional block. 2b. Instance where a drug prolongs the refractory period without significantly slowing conduction velocity. The impulse moving antegrade meets refractory tissue (shaded area) allowing for unidirectional block. A new reentrant tachycardia may result. B. Mechanism of reentry and proarrhythmia. a. Functionally determined (leading circle) reentrant circuit. This model should be contrasted with anatomic reentry; here the circuit is not fixed (it does not necessarily move around an anatomic obstacle) and there is no excitable gap. All tissue inside is held continuously refractory. b. Instance where a drug prolongs the refractory period without significantly slowing conduction velocity. The tachycardia may terminate or slow in rate as shown as a consequence of a greater circuit length. The dashed lines represent the original reentrant circuit prior to drug treatment. c. Instance where a drug slows conduction velocity without significantly prolonging the refractory period (i.e., class Ic antiarrhythmic drugs) and accelerates the tachycardia. The tachycardia rate may increase (proarrhythmia) as shown as a consequence of a shorter circuit length. The dashed lines represent the original reentrant circuit prior to drug treatment. (Reproduced with permission from McCollam PL, Parker RB, Beckman KJ, et al. Proarrhythmia: A paradoxic response to antiarrhythmic agents. Pharmacotherapy 1989;9:146.)
What causes reentry to become clinically manifest? Reentrant foci may occur at any level of the conduction system: within the branches of the specialized atrial conduction system, within the Purkinje network, and even within portions of the SA and AV nodes. The anatomy of the Purkinje system appears to provide a suitable substrate for the formation of microreentrant loops and is often used as a model to facilitate the understanding of reentry concepts (Fig. 8-4). Of course, because reentry does not usually occur in normal, healthy conduction tissue, various forms of heart disease or conduction abnormalities must usually be present before reentry becomes manifest. In other words, the various forms of heart disease (e.g., coronary artery disease [CAD], left ventricular [LV] dysfunction) can result in changes in conduction in the pathways of a suitable reentrant substrate. An often-used example is reentry occurring as a consequence of ischemic or hypoxic damage: with inadequate cellular oxygen, cardiac tissue resorts to anaerobic glycolysis for adenosine triphosphate production. As high-energy phosphate concentration diminish, the activity of the transmembrane ion pumps declines and RMP rises. This rise in RMP causes inactivation in the voltage-dependent sodium channel, and the tissue begins to assume slow conduction characteristics. If changes in conduction parameters occur in a discordant manner due to varying degrees of ischemia or hypoxia, then a reentry circuit may become manifest. Furthermore, an ischemic, dying cell liberates intracellular potassium, which also causes a rise in RMP. In other cases, reentry may occur as a consequence of anatomic or functional variants in the normal conduction system. For instance, patients may possess two (instead of one) conduction pathways near or within the AV node, or have an anomalous extranodal AV pathway that possesses different electrophysiologic characteristics from the normal AV nodal pathway. Reentry in these cases may occur within the AV node or encompass both atrial and ventricular tissues. Reentrant tachycardias have the following characteristics: (a) the onset of the tachycardia is usually related to an initiating event (i.e., premature beat); (b) the initiating beat is usually different in morphology from subsequent beats of the tachycardia; (c) the initiation of the tachycardia is usually possible with programmed cardiac stimulation; and (d) the initiation and termination of the tachycardia is usually abrupt without an acceleration or deceleration phase. There are many examples of reentrant tachycardias, including atrial fibrillation (AF), atrial flutter, AV nodal or AV reentrant tachycardia, and recurrent VT.
ANTIARRHYTHMIC DRUGS
In a theoretical sense, drugs may have antiarrhythmic activity by directly altering conduction in several ways. First, a drug may depress the automatic properties of abnormal pacemaker cells. A drug may do this by decreasing the slope of phase 4 depolarization and/or by elevating threshold potential. If the rate of spontaneous impulse generation of the abnormally automatic foci becomes less than that of the SA node, normal cardiac rhythm can be restored. Second, drugs may alter the conduction characteristics of the pathways of a reentrant loop.1,2 A drug may facilitate conduction (shorten refractoriness) in the area of unidirectional block, allowing antegrade conduction to proceed. On the other hand, a drug may further depress conduction (prolong refractoriness) either in the area of unidirectional block or in the pathway with slowed conduction and a relatively shorter refractory period. If refractoriness is prolonged in the area of unidirectional block, retrograde propagation of the impulse is not permitted, causing a “bidirectional” block. In the anatomic model, if refractoriness is prolonged in the pathway with slow conduction, antegrade conduction of the impulse is not permitted. In either case, drugs that reduce the discordance and cause uniformity in conduction properties of the two pathways may suppress the reentrant substrate. In the functionally determined model, if refractoriness is prolonged without significantly slowing conduction velocity, the tachycardia may terminate or slow in rate as a consequence of a greater circuit length (Fig. 8-4). There are other theoretical ways to stop reentry: a drug may eliminate the critically timed premature impulse that triggers reentry, a drug may slow conduction velocity to such an extent that conduction is extinguished, or a drug may reverse the underlying form of heart disease that was responsible for the conduction abnormalities that led to the arrhythmia (i.e., “reverse remodeling”).
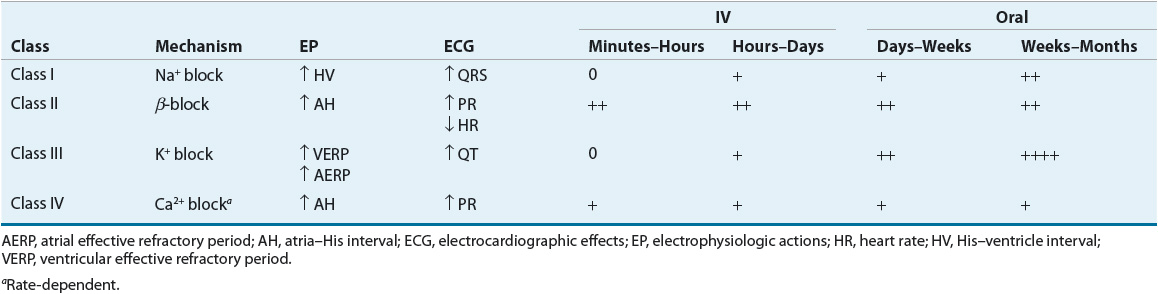
AADs have specific electrophysiologic actions that alter cardiac conduction in patients with or without heart disease. These actions form the basis of grouping AADs into specific categories based on their electrophysiologic actions in vitro. Vaughan Williams proposed the most frequently used classification system (Table 8-1).2 This classification has been criticized for the following reasons: (a) it is incomplete and does not allow for the classification of drugs such as digoxin or adenosine; (b) it is not pure, and many agents have properties of more than one class of drugs; (c) it does not incorporate drug characteristics such as mechanisms of tachycardia termination/prevention, clinical indications, or side effects; and (d) drugs become “labeled” within a class, although they may be distinct in many regards.3 These criticisms formed the basis for an attempt to reclassify AADs based on a variety of basic and clinical characteristics (called the Sicilian Gambit3). Nonetheless, the Vaughan Williams classification remains the most frequently used despite many proposed modifications and alternative systems.
TABLE 8-1 Classification of Antiarrhythmic Drugs
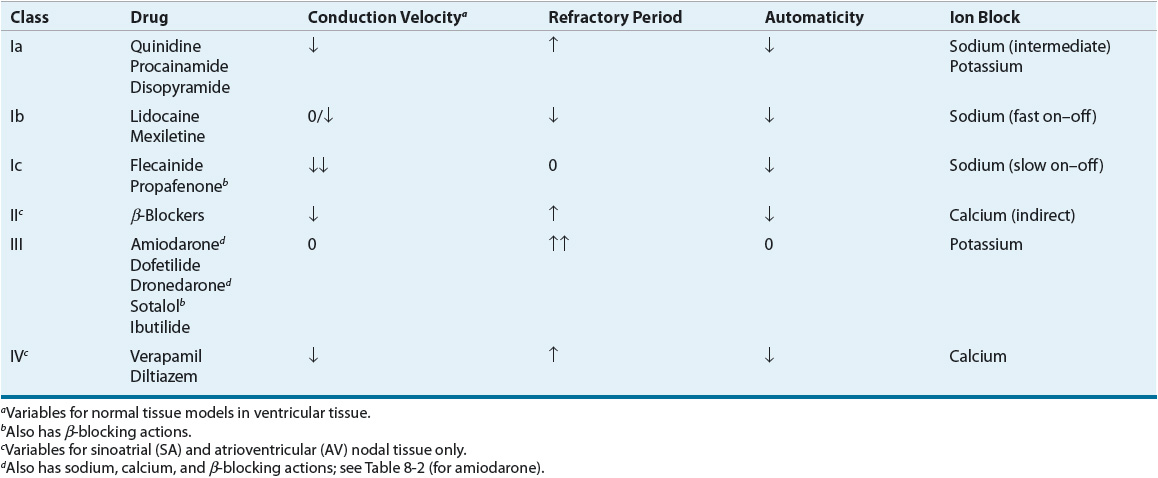
The class Ia AADs, quinidine, procainamide, and disopyramide, slow conduction velocity, prolong refractoriness, and decrease the automatic properties of sodium-dependent (normal and diseased) conduction tissue. Although class Ia AADs are primarily considered sodium channel blockers, their electrophysiologic actions can also be attributed to blockade of potassium channels. In reentrant tachycardias, these drugs generally depress conduction and prolong refractoriness, theoretically transforming the area of unidirectional block into a bidirectional block. Clinically, class Ia drugs are broad-spectrum AADs that are effective for both supraventricular and ventricular arrhythmias. Procainamide is only available in the IV formulation as all of its oral formulations have been discontinued.
The class Ib AADs, lidocaine, mexiletine, and phenytoin, were historically categorized separately from quinidine-like drugs. This was a result of early work demonstrating that lidocaine had distinctly different electrophysiologic actions. In normal tissue models, lidocaine generally facilitates actions on cardiac conduction by shortening refractoriness and having little effect on conduction velocity. Thus, it was postulated that these agents could improve antegrade conduction, eliminating the area of unidirectional block. Of course, arrhythmias do not usually arise from normal tissue, leading investigators to study the actions of lidocaine and phenytoin in ischemic and hypoxic tissue models. Interestingly, studies have shown these drugs to possess class Ia quinidine-like properties in diseased tissues. Therefore, it is probable that lidocaine acts in a similar fashion to the class Ia AADs (i.e., prolongs refractoriness in diseased ischemic tissues leading to bidirectional block in a reentrant circuit). Lidocaine and similar agents have accentuated effects in ischemic tissue caused by the local acidosis and potassium shifts that occur during cellular hypoxia. Changes in pH alter the time that local anesthetics occupy the sodium channel receptor, thereby affecting the agent’s electrophysiologic actions. In addition, the intracellular acidosis that ensues as a consequence of ischemia could cause lidocaine to become “trapped” within the cell, allowing increased access to the receptor. The class Ib AADs are considerably more effective in ventricular arrhythmias than supraventricular arrhythmias. As a group, these drugs are relatively weak sodium channel antagonists (at normal stimulation rates).
The class Ic AADs, propafenone and flecainide, are extremely potent sodium channel blockers, profoundly slowing conduction velocity while leaving refractoriness relatively unaltered. The class Ic AADs theoretically eliminate reentry by slowing conduction to a point where the impulse is extinguished and cannot propagate further. Although the class Ic AADs are effective for both ventricular and supraventricular arrhythmias, their use for ventricular arrhythmias has been limited by the risk of proarrhythmia.
Class I AADs are grouped together because of their common action in blocking sodium conductance. The receptor site for these AADs is probably inside the sodium channel so that, in effect, the drug plugs the pore. The AAD may gain access to the receptor either via the intracellular space through the membrane lipid bilayer or directly through the channel. Several principles are inherent in antiarrhythmic sodium channel receptor theories4:
1. Class I AADs have predominant affinity for a particular state of the channel (e.g., during activation or inactivation). For example, lidocaine and flecainide block sodium current primarily when the cell is in the inactivated state, whereas quinidine is predominantly an open (or activated)-channel blocker.
2. Class I AADs have specific binding and unbinding characteristics to the receptor. For example, lidocaine binds to and dissociates from the channel receptor quickly (“fast on–off”) but flecainide has very “slow on–off” properties. This explains why flecainide has such potent effects on slowing ventricular conduction, whereas lidocaine has little effect on normal tissue (at normal heart rates). In general, the class Ic AADs are “slow on–off,” the class Ib AADs are “fast on–off,” and the class Ia AADs are intermediate in their binding kinetics.
3. Class I AADs possess rate dependence (i.e., sodium channel blockade and slowed conduction are greatest at fast heart rates and least during bradycardia). For “slow on–off” drugs, sodium channel blockade is evident at normal rates (60 to 100 beats/min), but for “fast on–off” agents, slowed conduction is only apparent at fast heart rates.
4. Class I AADs (except phenytoin) are weak bases with a pKa >7 and block the sodium channel in their ionized form. Consequently, pH will alter these actions: acidosis accentuates and alkalosis diminishes sodium channel blockade.
5. Class I AADs appear to share a single receptor site in the sodium channel. It should be noted, however, that a number of class I AADs have other electrophysiologic properties. For instance, quinidine has potent potassium channel blocking activity (manifests predominantly at low concentrations) as does N-acetylprocainamide (manifests predominantly at high concentrations), the primary metabolite of procainamide. Additionally propafenone has β-blocking actions.
These principles are important in understanding additive drug combinations (e.g., quinidine and mexiletine), antagonistic combinations (e.g., flecainide and lidocaine), and potential antidotes to excess sodium channel blockade (sodium bicarbonate). They also explain a number of clinical observations, such as why lidocaine-like drugs are relatively ineffective for supraventricular tachycardia. The class Ib AADs are “fast on–off,” inactivated sodium channel blockers; atrial cells, however, have a very brief inactivated phase relative to ventricular tissue.
The β-blockers are classified as class II AADs. For the most part, the clinically relevant acute antiarrhythmic mechanisms of the β-blockers result from their antiadrenergic actions. Because the SA and AV nodes are heavily influenced by adrenergic innervation, β-blockers would be most useful in tachycardias in which these nodal tissues are abnormally automatic or are a portion of a reentrant loop. These drugs are also helpful in slowing ventricular response in atrial arrhythmias (e.g., AF) by their effects on the AV node. Furthermore, some tachycardias are exercise-related or precipitated by states of high sympathetic tone (perhaps through triggered activity), and β-blockers may be useful in these instances. β-Adrenergic stimulation results in increased conduction velocity, shortened refractoriness, and increased automaticity of the nodal tissues; β-blockers will antagonize these effects. In the nodal tissues, β-blockers interfere with calcium entry into the cell by altering catecholamine-dependent channel integrity and gating kinetics. In sodium-dependent atrial and ventricular tissues, β-blockers shorten repolarization somewhat but otherwise have little direct effect. The antiarrhythmic properties of β-blockers observed with long-term, chronic therapy in patients with heart disease are less well understood. Although it is clear that β-blockers decrease the likelihood of SCD (presumably arrhythmic death) after myocardial infarction (MI), the mechanism for this benefit remains unclear but may relate to the complex interplay of changes in sympathetic tone, damaged myocardium, and ventricular conduction. In patients with heart failure (HF), drugs such as β-blockers, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers may prevent arrhythmias such as AF by attenuating the structural and/or electrical remodeling process in the myocardium.5,6
The class III AADs include those agents that specifically prolong refractoriness in atrial and ventricular tissues. This class includes amiodarone, dronedarone, sotalol, ibutilide, and dofetilide; these drugs share the common effect of delaying repolarization by blocking potassium channels. Amiodarone and sotalol are effective in most supraventricular and ventricular arrhythmias. Amiodarone displays electrophysiologic characteristics of all four Vaughan Williams classes; it is a sodium channel blocker with relatively “fast on–off” kinetics, has nonselective β-blocking actions, blocks potassium channels, and also has a small degree of calcium channel blocking activity (Table 8-2). At normal heart rates and with chronic use, its predominant effect is to prolong repolarization. With IV administration, its onset is relatively quick (unlike the oral form) and β-blockade predominates initially. Theoretically, amiodarone, like class I AADs, may interrupt the reentrant substrate by transforming an area of unidirectional block into an area of bidirectional block. However, electrophysiologic studies using programmed cardiac stimulation imply that amiodarone may leave the reentrant loop intact. The impressive effectiveness of amiodarone coupled with its low proarrhythmic potential has challenged the notion that selective ion channel blockade by AADs is preferable. Sotalol is a potent inhibitor of outward potassium movement during repolarization and also possesses nonselective β-blocking actions. Unlike amiodarone and sotalol, dronedarone, ibutilide, and dofetilide are only approved for the treatment of supraventricular arrhythmias. Both ibutilide (only available IV) and dofetilide (only available orally) can be used for the acute conversion of AF or atrial flutter to SR. Dofetilide can also be used to maintain SR in patients with AF or atrial flutter of longer than 1 week’s duration who have been converted to sinus rhythm (SR). Dronedarone is approved to reduce the risk of cardiovascular hospitalization in patients with a history of paroxysmal or persistent AF. Although structurally related to amiodarone, dronedarone’s structure has been modified through the addition of a methylsulfonyl group and the removal of iodine. Dronedarone is also similar to amiodarone in exhibiting electrophysiologic characteristics of all four Vaughan Williams classes (sodium channel blocker with relatively “fast on–off” kinetics, nonselective β-blocker, potassium channel blocker, and calcium channel antagonist).
TABLE 8-2 Time Course and Electrophysiologic Effects of Amiodarone
There are a number of different potassium channels that function during normal conduction; all approved class III AADs inhibit the delayed rectifier current (IK) responsible for phase 2 and phase 3 repolarization. Subcurrents make up IK: an ultrarapid component (IKur), a rapid component (IKr), and the slow component (IKs). N-Acetylprocainamide, sotalol, ibutilide, and dofetilide selectively block IKr, whereas amiodarone and dronedarone block both IKr and IKs. New drugs that selectively block IKur (found predominantly in the atrium but not ventricle) are being investigated for supraventricular arrhythmias. The clinical relevance of selectively blocking components of the delayed rectifier current remains to be determined. Potassium channel blockers (particularly those with selective IKr blocking properties) display “reverse use dependence” (i.e., their effects on repolarization are greatest at low heart rates). Sotalol and drugs like it also appear to be much more effective in preventing ventricular fibrillation (VF) (in dog models) than the traditional sodium channel blockers. They also decrease defibrillation threshold in contrast to class I AADs, which tend to increase this parameter. This feature could be important in patients with ICDs, as concurrent therapy with class I AADs may require more energy for successful cardioversion or may render the ICD ineffective in terminating the ventricular arrhythmia. The Achilles’ heel of all class III AADs is an extension of their underlying ionic mechanism (i.e., by blocking potassium channels and delaying repolarization, they may also cause proarrhythmia in the form of TdP by provoking EADs).
The nondihydropyridine (non-DHP) CCBs, verapamil and diltiazem, are categorized as class IV AADs. At least two types of calcium channels are operative in SA and AV nodal tissues: an L-type channel and a T-type channel. Both L-type channel blockers (verapamil and diltiazem) and selective T-type channel blockers (mibefradil was previously approved but withdrawn from the market) will slow conduction, prolong refractoriness, and decrease automaticity (e.g., due to EADs or LADs) of the calcium-dependent tissue in the SA and AV nodes. Therefore, these agents are effective in automatic or reentrant tachycardias, which arise from or use the SA or AV nodes. In supraventricular arrhythmias (e.g., AF), these drugs can slow ventricular response by slowing AV nodal conduction. Furthermore, because calcium entry seems to be integral to exercise-related tachycardias and/or tachycardias caused by some forms of triggered automaticity, these agents may be effective in the treatment of these types of arrhythmias. The DHP CCBs (e.g., nifedipine) do not have significant antiarrhythmic activity because a reflex increase in sympathetic tone caused by vasodilation counteracts their direct negative dromotropic action.
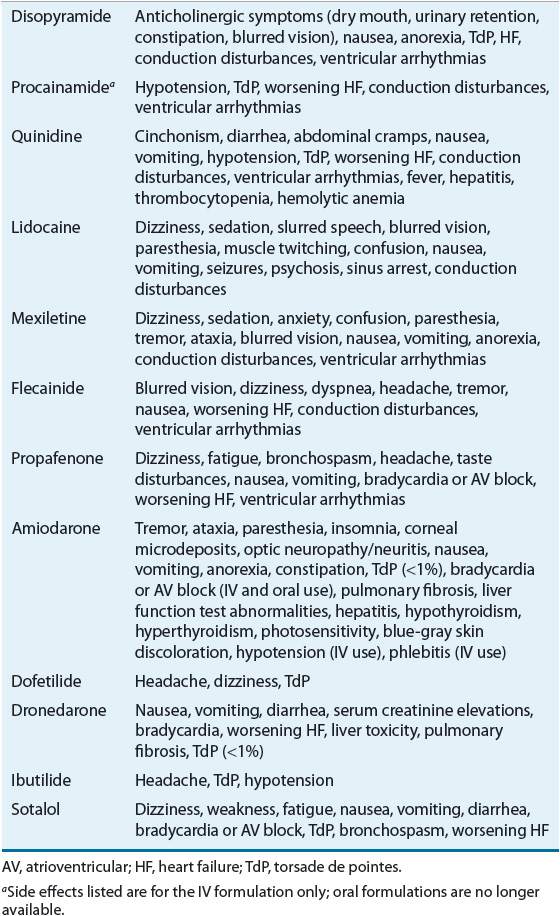
All AADs currently available have an impressive side effect profile (Table 8-3). A considerable percentage of patients cannot tolerate long-term therapy with these drugs and will have to discontinue therapy because of side effects. ![]() Flecainide, propafenone, and disopyramide may precipitate congestive HF in a significant number of patients with underlying LV systolic dysfunction; consequently, these drugs should be avoided in this patient population.7 The class Ib AAD, mexiletine, causes neurologic and/or gastrointestinal (GI) toxicity in a high percentage of patients. One of the most frightening side effects related to AADs is the aggravation of underlying ventricular arrhythmias or the precipitation of new (and life-threatening) ventricular arrhythmias.8
Flecainide, propafenone, and disopyramide may precipitate congestive HF in a significant number of patients with underlying LV systolic dysfunction; consequently, these drugs should be avoided in this patient population.7 The class Ib AAD, mexiletine, causes neurologic and/or gastrointestinal (GI) toxicity in a high percentage of patients. One of the most frightening side effects related to AADs is the aggravation of underlying ventricular arrhythmias or the precipitation of new (and life-threatening) ventricular arrhythmias.8
TABLE 8-3 Side Effects of Antiarrhythmic Drugs
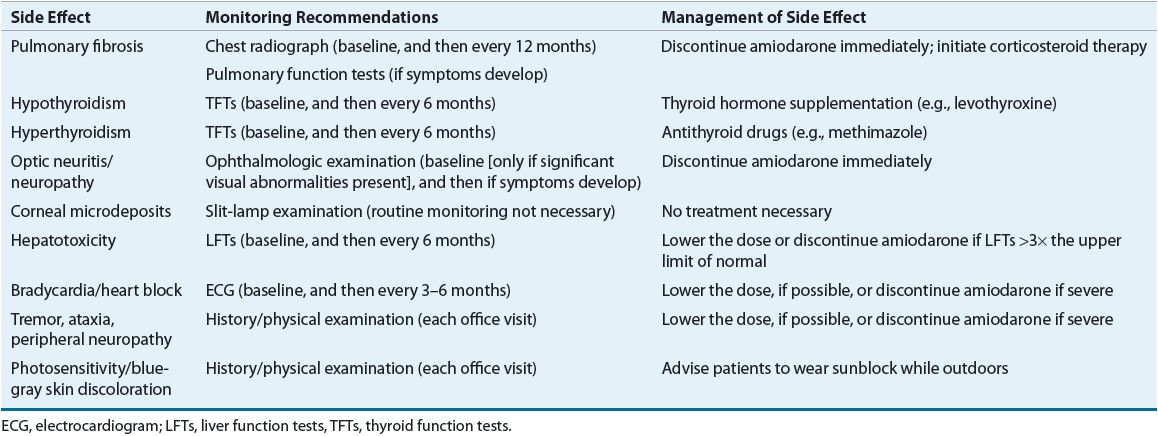
Amiodarone has assumed a prominent place in the treatment of both chronic and acute supraventricular and ventricular arrhythmias and is now the most commonly prescribed AAD.9 Once considered a drug of last resort, it is now the first AAD considered in many arrhythmias. Yet amiodarone is a peculiar and complex drug, displaying unusual pharmacologic effects, pharmacokinetics, dosing regimens, and multiorgan side effects. Amiodarone has an extremely long elimination half-life (greater than 50 days) and large volume of distribution; consequently, its onset of action with the oral form is delayed (days to weeks) despite the use of a loading regimen, and its effects persist for a long period (months) after discontinuation. Amiodarone is a substrate of the cytochrome P450 (CYP) 3A4 isoenzyme, a moderate inhibitor of many CYP isoenzymes (e.g., CYP2C9, CYP2D6, CYP3A4), and a P-glycoprotein inhibitor, all of which can result in the potential for numerous drug interactions. Amiodarone interacts with digoxin and warfarin and can significantly increase plasma concentrations of both drugs. By inhibiting P-glycoprotein, amiodarone can increase digoxin concentrations by approximately twofold; therefore, the digoxin dose should be empirically reduced by 50% when amiodarone is initiated. When amiodarone and warfarin are initiated concurrently, warfarin should be started at a dose of 2.5 mg daily. When amiodarone is initiated in a patient already receiving warfarin, the warfarin dose should be reduced by approximately 30%.10 Acute administration of amiodarone is usually well tolerated by patients, but severe organ toxicities may result with chronic use. Severe bradycardia (sometimes requiring pacing to allow the patient to remain on amiodarone), hyperthyroidism, hypothyroidism, peripheral neuropathy, GI discomfort, photosensitivity, and a blue-gray skin discoloration on exposed areas are common. Fulminant hepatitis (uncommon) and pulmonary fibrosis (5% to 10% of patients) have caused death.11,12 Although amiodarone can cause corneal microdeposits (usually do not affect vision) in virtually every patient, it has also been associated with the development of optic neuropathy/neuritis, which can lead to blindness. All of these side effects mandate close and continued monitoring (liver enzymes, thyroid function tests, eye examinations, chest radiographs, pulmonary function tests) and have led to a proliferation of “amiodarone clinics” designed just for patients receiving this drug on a chronic basis (Table 8-4). ![]() 13,14
13,14
The modifications to dronedarone’s chemical structure may confer an improved safety profile when compared with amiodarone. With the addition of a methylsulfonyl group and the deletion of the iodine moiety, dronedarone is less lipophilic than amiodarone; consequently, dronedarone is supposed to be less likely to accumulate in tissues and cause various organ toxicities. Dronedarone also has a considerably shorter half-life (∼24 hours) when compared with amiodarone, which allows for steady state to be achieved in 5 to 7 days, without the need for using loading doses. Like amiodarone, dronedarone is a substrate of the CYP3A isoenzyme and a moderate inhibitor of the CYP2D6 and CYP3A isoenzymes. Its use with potent CYP3A4 inhibitors or inducers should be avoided. Dronedarone may increase plasma concentrations of (S)-warfarin; therefore, the international normalized ratio (INR) should be closely monitored with concurrent use of these drugs. Dronedarone also inhibits P-glycoprotein and can increase digoxin concentrations by about 2.5-fold. Consequently, when concomitantly using dronedarone and digoxin, the digoxin dose should be empirically reduced by 50%. Additionally, dronedarone can increase dabigatran and rivaroxaban concentrations in patients with renal impairment. To minimize the risk of bleeding when concomitantly using dronedarone and dabigatran in this patient population, the dose of dabigatran should be reduced to 75 mg twice daily in those with moderate renal impairment (creatinine clearance [CrCl] 30 to 50 mL/min). The concomitant use of dronedarone and dabigatran should be avoided in patients with severe renal impairment (CrCl 15 to 30 mL/min). Rivaroxaban should only be used if the benefit outweighs the risk in patients receiving dronedarone who have a CrCl of 15 to 50 mL/min.
Table 8-5 summarizes the pharmacokinetics of the AADs and Table 8-6 lists recommended dosages of the oral dosage forms. Table 8-7 lists the dosing recommendations for the IV forms of various AADs.
Table 8-5 Pharmacokinetics of Antiarrhythmic Drugs
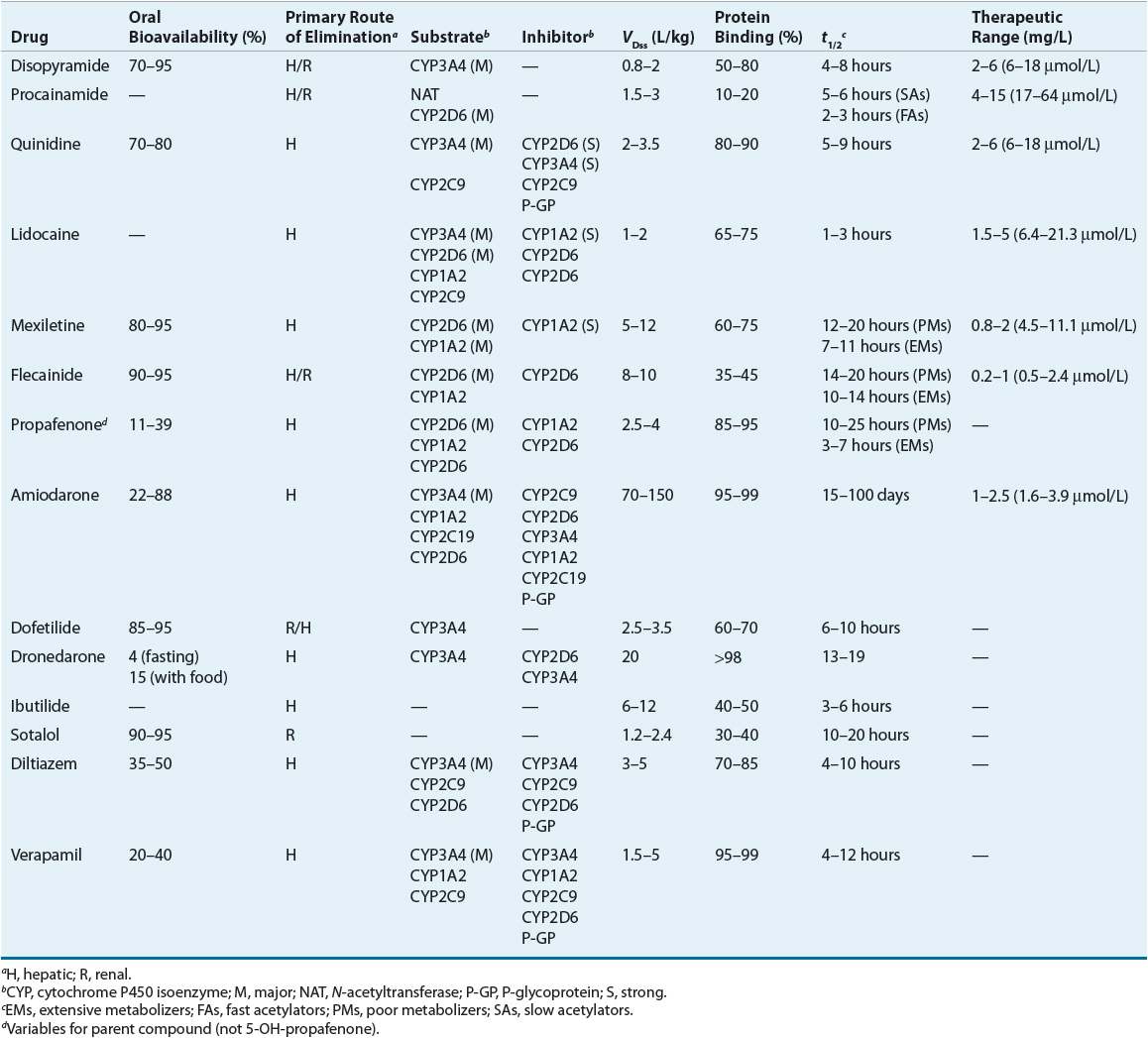
Table 8-6 Typical Maintenance Doses of Oral Antiarrhythmic Drugs
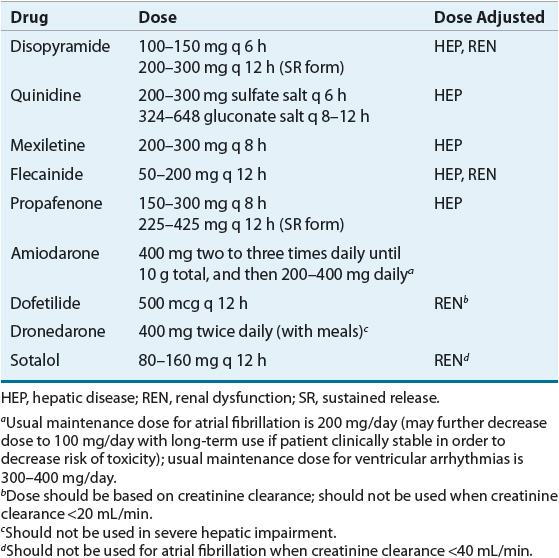
Table 8-7 IV Antiarrhythmic Dosing

SUPRAVENTRICULAR ARRHYTHMIAS
The common supraventricular tachycardias that often require drug treatment are: (a) AF or atrial flutter, (b) paroxysmal supraventricular tachycardia (PSVT), and (c) automatic atrial tachycardias. Other common supraventricular arrhythmias that usually do not require drug therapy include premature atrial complexes, wandering atrial pacemaker, sinus arrhythmia, and sinus tachycardia. As an example, premature atrial complexes rarely cause symptoms and never cause hemodynamic compromise; therefore, drug therapy is usually not indicated. Likewise, sinus tachycardia is usually the result of underlying metabolic or hemodynamic disorders (e.g., infection, dehydration, hypotension), and therapy should be directed at the underlying cause, not the tachycardia per se. Of course, there are exceptions to these suggestions. For example, sinus tachycardia may be deleterious in patients after cardiac surgery or MI. Therefore, AADs, such as β-blockers, may be indicated in these situations. Stated in another way, although many arrhythmias generally do not require therapy, clinical judgment and patient-specific variables play an important role in this decision. AF, atrial flutter, and PSVT tend to be the most common supraventricular arrhythmias seen in clinical practice; therefore, this discussion will focus only on these arrhythmias.
Atrial Fibrillation and Atrial Flutter
Mechanisms and Background
AF continues to be the most common sustained arrhythmia encountered in clinical practice, affecting between 2.7 and 6.1 million Americans.15 In the general population, the overall prevalence of AF is 0.4% to 1%, and this increases with age (e.g., approximately an 8% prevalence in patients >80 years old).16 The prevalence of AF also appears to increase as patients develop more severe HF, increasing from 4% in asymptomatic New York Heart Association (NYHA) class I patients to 50% in patients with NYHA class IV HF.17 With the aging population, improved survival in patients with HF, CAD, and hypertension, and the increased frequency of surgical procedures being performed, it is expected that the prevalence of AF will dramatically increase to an estimated 12 to 15 million by the year 2050.17 Based on data derived from the Framingham study cohort, the general lifetime risk for AF in men and women at least 40 years of age is estimated to be 1 in 4.18
AF and atrial flutter may present as a chronic, established tachycardia, an acute tachycardia, or a self-terminating, paroxysmal form. The following semantics and definitions are sometimes used specifically for AF16,19: acute AF (onset within 48 hours), paroxysmal AF (terminates spontaneously in <7 days), recurrent AF (two or more episodes), persistent AF (duration >7 days and does not terminate spontaneously), and permanent AF (does not terminate with attempts at pharmacologic or electrical cardioversion). AF is characterized by extremely rapid (atrial rate of 400 to 600 beats/min) and disorganized atrial activation. With this disorganized atrial activity, there is a loss of the contribution of synchronized atrial contraction (atrial kick) to forward cardiac output. Supraventricular impulses penetrate the AV conduction system in variable degrees resulting in an irregular activation of the ventricles and an irregularly irregular pulse. The AV junction will not conduct most of the supraventricular impulses, causing the ventricular response to be considerably slower (120 to 180 beats/min) than the atrial rate. It is sometimes stated that “AF begets AF,” that is, the arrhythmia tends to perpetuate itself. Long episodes are more difficult to terminate perhaps because of tachycardia-induced changes in atrial function (mechanical and/or electrical “remodeling”).
CLINICAL PRESENTATION Supraventricular Tachycardias
Atrial flutter occurs less frequently than AF but is similar in its precipitating factors, consequences, and drug therapy approach. This arrhythmia is characterized by rapid (atrial rate of 270 to 330 beats/min) but regular atrial activation. The slower and regular electrical activity results in a regular ventricular response that is in approximate factors of 300 beats/min (i.e., 1:1 AV conduction = ventricular rate of 300 beats/min; 2:1 AV conduction = ventricular rate of 150 beats/min; 3:1 AV conduction = ventricular rate of 100 beats/min). Atrial flutter may occur in two distinct forms (type I and type II). Type I flutter is the more common classic form with atrial rates of approximately 300 beats/min and the typical “sawtooth” pattern of atrial activation as shown by the surface ECG. Type II flutter tends to be faster, being somewhat of a hybrid between classic atrial flutter and AF. Although the ventricular response usually has a regular pattern with this arrhythmia, atrial flutter with varying degrees of AV block or that occur with episodes of AF (“fib-flutter”) can cause an irregular ventricular rate.
It is generally accepted that the predominant mechanism of AF and atrial flutter is reentry. AF appears to result from multiple atrial reentrant loops (or wavelets), whereas atrial flutter is caused by a single, dominant, reentrant substrate (counterclockwise circus movement in the right atrium around the tricuspid annulus). AF or atrial flutter usually occurs in association with various forms of structural heart disease (SHD) that cause atrial distension, including myocardial ischemia or infarction, hypertensive heart disease, valvular disorders such as mitral stenosis or mitral insufficiency, congenital abnormalities such as septal defects, dilated or hypertrophic cardiomyopathy, and obesity. Disorders that cause right atrial stretch and are associated with AF or atrial flutter include acute pulmonary embolus and chronic lung disease resulting in pulmonary hypertension and cor pulmonale. AF may also occur in association with states of high adrenergic tone such as thyrotoxicosis, surgery, alcohol withdrawal, sepsis, and excessive physical exertion. AF that develops in the absence of clinical, electrocardiographic, radiographic, and echocardiographic evidence of SHD is defined as lone AF. Other states in which patients are predisposed to episodes of AF are the presence of an anomalous AV pathway (i.e., Kent’s bundle) and sinus node dysfunction (i.e., sick sinus syndrome).
Patients with AF or atrial flutter may experience the entire range of symptoms associated with other supraventricular tachycardias, although syncope as a presenting symptom is uncommon. Because atrial kick is lost with the onset of AF, patients with LV systolic or diastolic dysfunction may develop worsening signs and symptoms of HF as they often depend on the contribution of their atrial kick to maintain an adequate cardiac output. Thromboembolic events, resulting from atrial stasis and poorly adherent mural thrombi, are an additional complication of AF. Of course, the most devastating complication in this regard is the occurrence of an embolic stroke. The average rate of ischemic stroke in patients with AF who are not receiving antithrombotic therapy is approximately 5% per year.20,21 Stroke can precede the onset of documented AF, probably as a result of undetected paroxysms prior to the onset of established AF. The risk of stroke significantly increases with age, with the annual attributable risk increasing from 1.5% in individuals 50 to 59 years of age to almost 24% in those 80 to 89 years of age.20 Patients with concomitant AF and rheumatic heart disease are at particularly high risk for stroke, with their risk being increased 17-fold compared with patients in SR.20 Other risk factors for stroke identified from recent trials are previous ischemic stroke, transient ischemic attack, or other systemic embolic event; age >75 years; moderate or severe LV systolic dysfunction and/or congestive HF; hypertension; and diabetes.20 The risk of stroke in patients with only atrial flutter has been traditionally believed to be low, prompting some to recommend only aspirin for prevention of thromboembolism in this particular patient population. However, because patients with atrial flutter may also intermittently have episodes of AF, this patient population may also be at risk for a thromboembolic event. Although the role of antithrombotic therapy in patients with atrial flutter has not been adequately studied in clinical trials, the most recent guidelines suggest that the same risk stratification scheme and antithrombotic recommendations used in patients with AF should also be applied to those with atrial flutter.20
Management
The traditional approach to the treatment of AF can be organized into several sequential goals. First, evaluate the need for acute treatment (usually administering drugs that slow ventricular rate). Next, contemplate methods to restore SR, taking into consideration the risks (e.g., thromboembolism). Lastly, consider ways to prevent the long-term complications of AF such as arrhythmia recurrence and thromboembolism. ![]() One of the biggest controversies in the management of AF is whether restoring and maintaining SR is a desirable goal for all patients. A review of the management of AF and atrial flutter, including a discussion of this controversy, follows, organized according to the goals already outlined. Figure 8-5 shows an algorithm for the management of AF and atrial flutter. In addition, Table 8-8 summarizes the recommendations for pharmacologically controlling ventricular rate and restoring and maintaining SR from the most recent AF guidelines developed by the American College of Cardiology (ACC)/American Heart Association (AHA)/European Society of Cardiology (ESC).16,22
One of the biggest controversies in the management of AF is whether restoring and maintaining SR is a desirable goal for all patients. A review of the management of AF and atrial flutter, including a discussion of this controversy, follows, organized according to the goals already outlined. Figure 8-5 shows an algorithm for the management of AF and atrial flutter. In addition, Table 8-8 summarizes the recommendations for pharmacologically controlling ventricular rate and restoring and maintaining SR from the most recent AF guidelines developed by the American College of Cardiology (ACC)/American Heart Association (AHA)/European Society of Cardiology (ESC).16,22
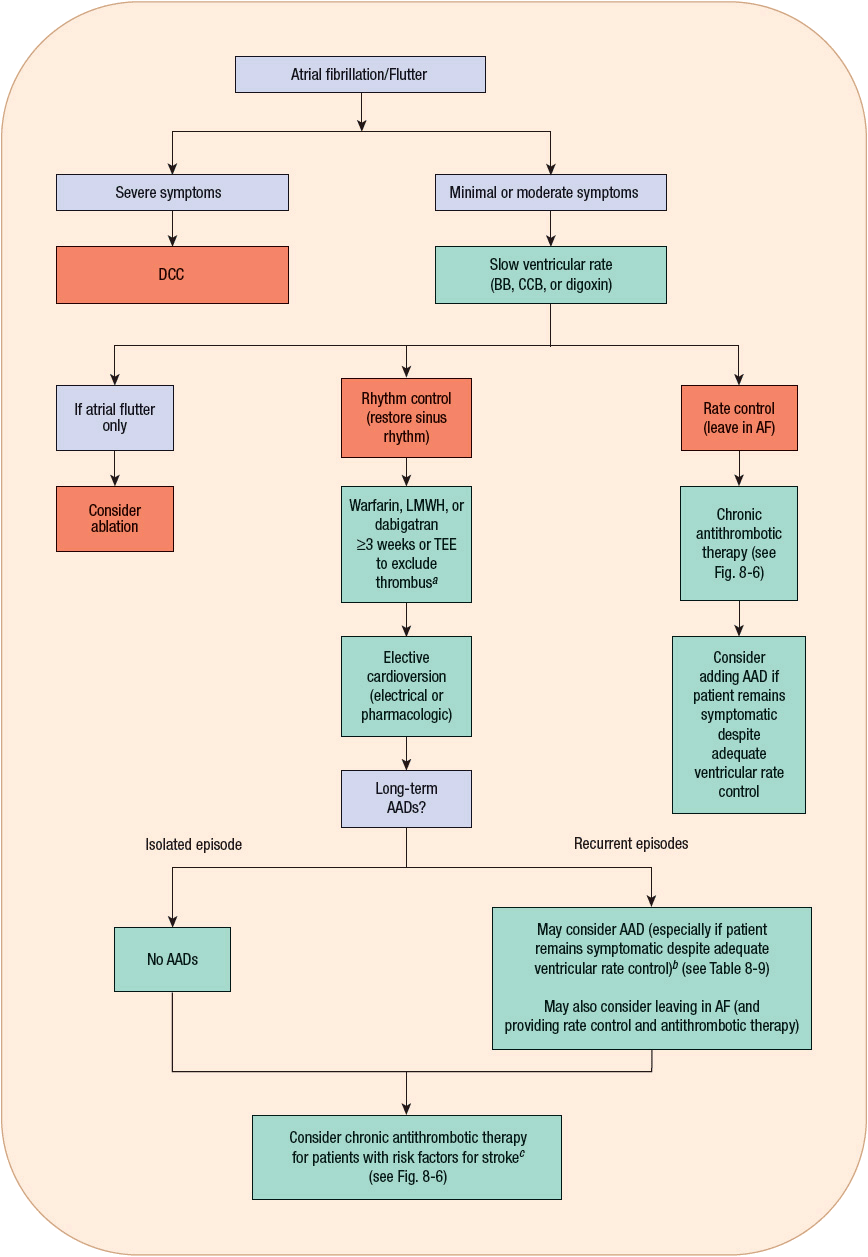
FIGURE 8-5 Algorithm for the treatment of AF and atrial flutter. aIf AF <48 hours, anticoagulation prior to cardioversion is unnecessary; may consider TEE if patient has risk factors for stroke. bAblation may be considered for patients who fail or do not tolerate ≥1 AAD. cChronic antithrombotic therapy should be considered in all patients with AF and risk factors for stroke regardless of whether or not they remain in sinus rhythm. (AAD, antiarrhythmic drug; AF, atrial fibrillation; BB, β-blocker; CCB, calcium channel blocker [i.e., verapamil or diltiazem]; DCC, direct current cardioversion; LMWH, low-molecular-weight heparin; TEE, transesophageal echocardiogram.)
Table 8-8 Evidence-Based Pharmacologic Treatment Recommendations for Controlling Ventricular Rate, Restoring Sinus Rhythm, and Maintaining Sinus Rhythm in Patients with Atrial Fibrillation
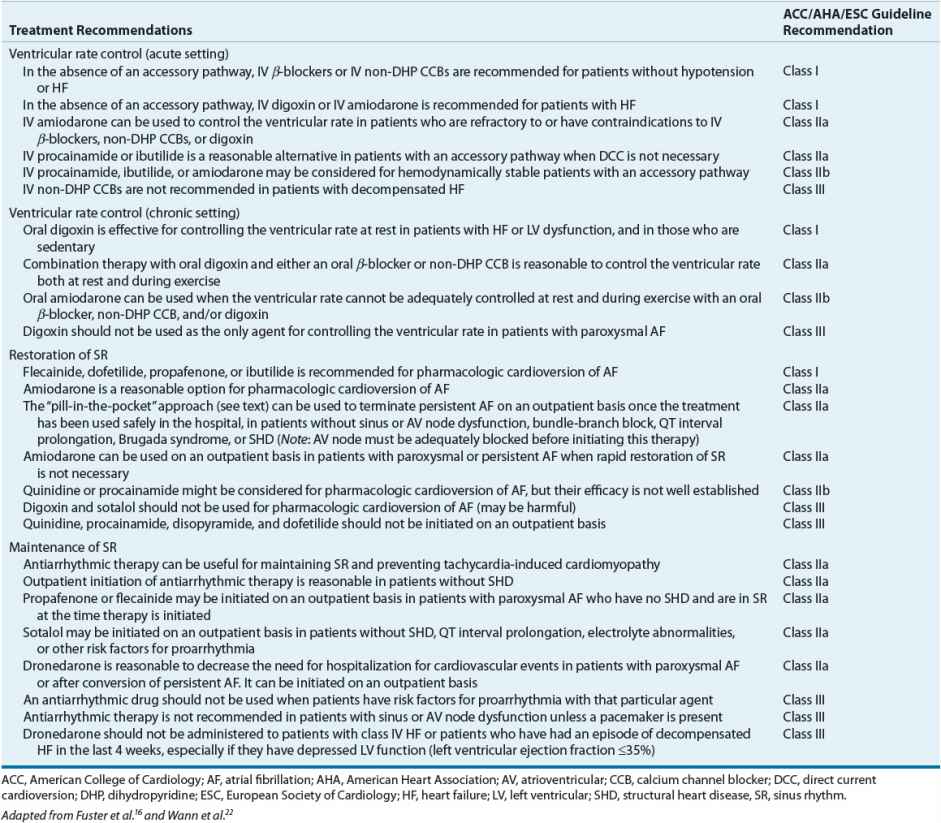
Acute Treatment First, consider the patient with new-onset, symptomatic AF or atrial flutter. Although uncommon, patients may present with signs and/or symptoms of hemodynamic instability (e.g., severe hypotension, angina, or pulmonary edema), which qualifies as a medical emergency. In these situations, direct current cardioversion (DCC) is indicated as first-line therapy in an attempt to immediately restore SR (without regard to the risk of thromboembolism). Atrial flutter often requires relatively low energy levels of countershock (i.e., 50 J), whereas AF often requires higher energy levels (i.e., greater than 200 J).
If patients are hemodynamically stable, there is no emergent need to restore SR. Instead, the focus should be directed toward controlling the patient’s ventricular rate. Achieving adequate ventricular rate control should be a treatment goal for all patients with AF. To achieve this goal, drugs that slow conduction and increase refractoriness in the AV node (e.g., β-blockers, non-DHP CCBs, or digoxin) should be used as initial therapy. Although loading doses of digoxin have been historically recommended as first-line treatment to slow ventricular rate, use of this drug for this purpose, especially in patients with normal LV systolic function (left ventricular ejection fraction [LVEF] >40%), has declined.9 In this patient population, IV β-blocker (propranolol, metoprolol, esmolol), diltiazem, or verapamil is preferred. A few of the potential reasons for the declining use of digoxin in this patient population are its relatively slow onset and its inability to control the ventricular rate during exercise. Although an initial decrease in the ventricular rate can sometimes be observed within 1 hour of IV administration of digoxin, full control (heart rate <80 beats/min at rest and <100 beats/min during exercise) is usually not achieved for 24 to 48 hours. In addition, digoxin tends to be ineffective for controlling ventricular rate under conditions of increased sympathetic tone (i.e., surgery, thyrotoxicosis) because it slows AV nodal conduction primarily through vagotonic mechanisms. In contrast, IV β-blockers and non-DHP CCBs have a relatively quick onset and can effectively control the ventricular rate at rest and during exercise. β-Blockers are also effective for controlling ventricular rate under conditions of increased sympathetic tone.
Based on the most recent guidelines for the treatment of AF, the selection of a drug to control ventricular rate in the acute setting should be primarily based on the patient’s LV function.16 In patients with normal LV function (LVEF >40%), IV β-blocker, diltiazem, or verapamil is recommended as first-line therapy to control ventricular rate.16 All of these drugs have proven efficacy in controlling the ventricular rate in patients with AF. Propranolol and metoprolol can be administered as intermittent IV boluses, whereas esmolol (because of its very short half-life of 5 to 10 minutes) must be administered as a series of loading doses followed by a continuous infusion. Likewise, because control of ventricular rate can be transient with a single bolus, verapamil or diltiazem can be given as an initial IV bolus followed by a continuous infusion.23 These continuous infusions can be adjusted in monitored settings to the desired ventricular response (e.g., acutely <100 beats/min). In situations where AF or atrial flutter is precipitated by states of increased sympathetic tone (i.e., surgery, thyrotoxicosis), IV β-blockers can be highly effective and should be considered first.
In patients with LV dysfunction (LVEF ≤40%), both IV diltiazem and verapamil should be avoided because of their potent negative inotropic effects. IV β-blockers should be used with caution in this patient population and should be avoided if patients are in the midst of an episode of decompensated HF. In those patients who are having an exacerbation of HF symptoms, IV administration of either digoxin or amiodarone should be used as first-line therapy to achieve ventricular rate control.16 IV amiodarone can also be used in patients who are refractory to or have contraindications to β-blockers, non-DHP CCBs, and digoxin.16 However, clinicians should be aware that the use of amiodarone for controlling ventricular rate may also stimulate the conversion of AF to SR and place the patient at risk for a thromboembolic event, especially if the AF has persisted for at least 48 hours or is of unknown duration.
Patients may present with a slow ventricular response (in the absence of AV nodal blocking drugs) and thus do not require therapy with β-blockers, non-DHP CCBs, or digoxin. This type of presentation should alert the clinician to the possibility of preexisting SA or AV nodal conduction disease such as sick sinus syndrome. In these patients, DCC should not be attempted without a temporary pacemaker in place.
Restoration of Sinus Rhythm? After treatment with AV nodal blocking drugs and a subsequent decrease in the ventricular rate, the patient should be evaluated for the possibility of restoring SR if AF persists. Within the context of this evaluation, several factors should be considered. First, many patients spontaneously convert to SR without intervention, obviating the need for therapy to achieve this goal. For instance, AF occurs frequently as a complication of cardiac surgery and often spontaneously reverts to SR without therapy. Second, restoring SR is not a necessary or realistic goal in some patients. The results of six landmark clinical trials (Pharmacological Intervention in Atrial Fibrillation [PIAF], Rate Control Versus Electrical Cardioversion for Persistent Atrial Fibrillation [RACE], Atrial Fibrillation Follow-Up Investigation of Rhythm Management [AFFIRM], Strategies of Treatment of Atrial Fibrillation [STAF], How to Treat Chronic Atrial Fibrillation [HOT-CAFE], and Atrial Fibrillation and Congestive Heart Failure [AF-CHF]) have shed significant light on the comparative efficacy of rate-control (controlling ventricular rate; patient remains in AF) and rhythm-control (restoring and maintaining SR) treatment strategies in patients with AF.24–29 The AFFIRM trial is the largest rate-control versus rhythm-control study to be conducted to date in patients with AF.26 In this trial, patients with AF and at least one risk factor for stroke were randomized to either a rate-control or a rhythm-control group. Rate-control treatment involved AV nodal blocking drugs (digoxin, β-blockers, and/or non-DHP CCBs) first, and then nonpharmacologic treatment (AV nodal ablation with pacemaker implantation), if necessary. All patients in this group were anticoagulated with warfarin to achieve an INR of 2 to 3. In the rhythm-control group, class I or III AADs were used to maintain SR. The choice of AAD therapy was left up to each patient’s physician; however, by the end of the trial, more than 60% of patients had received at least one trial of amiodarone and approximately 40% of patients had received at least one trial of sotalol. In this group, anticoagulation was encouraged but could be discontinued if SR had been maintained for at least 4 weeks. After a mean follow-up period of 3.5 years, overall mortality was not statistically different between the two strategies but tended (P = 0.08) to be higher in the rhythm-control group. The results of the PIAF, RACE, STAF, and HOT-CAFE trials were consistent with those of the AFFIRM trial.24,25,27,28 In addition, a meta-analysis of the data from all of these trials demonstrated no significant difference in overall mortality between rate-control and rhythm-control strategies, which persisted even when the results from the AFFIRM trial were excluded from this analysis.30
Even though the results of the PIAF, RACE, STAF, HOT-CAFE, and AFFIRM trials collectively demonstrate that a rate-control strategy is a viable alternative to a rhythm-control strategy in patients with persistent AF, a significant limitation of these results is that they cannot be applied to patients with HF because only a small proportion of patients enrolled in these trials had LV systolic dysfunction. The AF-CHF trial was conducted to specifically evaluate the safety and efficacy of rate-control and rhythm-control strategies in patients with systolic HF.29 In this trial, patients with an LVEF ≤35%, a history of HF (defined as NYHA class II to IV HF within the last 6 months, NYHA class I HF with a hospitalization for HF during the previous 6 months, or an LVEF ≤25%), and a history of AF were randomized to either a rate-control or a rhythm-control group. Rate-control treatment involved concomitant therapy with a β-blocker and digoxin first, and then nonpharmacologic treatment (AV nodal ablation with pacemaker implantation), if necessary. In the rhythm-control group, amiodarone was the preferred AAD, whereas sotalol and dofetilide were considered alternatives (most of the patients ultimately received amiodarone). If patients in this group did not convert to SR within 6 weeks, electrical cardioversion was performed. Anticoagulation was recommended for all patients in both treatment groups. After a mean follow-up period of 37 months, no significant difference was observed between the treatment groups with regard to the primary end point of death from cardiovascular causes. Patients in the rhythm-control group tended to have more hospitalizations, primarily due to repeated cardioversions and adjustment of AAD therapy, compared with patients in the rate-control group; however, this difference was not statistically significant (P = 0.06). It is important to note that the results of this trial should not be applied to patients with HF and preserved LV function (i.e., diastolic HF). Nevertheless, the results of this trial are generally consistent with those of the PIAF, RACE, AFFIRM, STAF, and HOT-CAFE trials and suggest that a rhythm-control strategy does not confer any advantage over a rate-control strategy in patients with AF and systolic HF.
Clearly, these important findings temper the old approach of aggressively attempting to maintain SR. Because a rhythm-control strategy does not offer any significant advantage over a rate-control strategy in the management of patients with persistent or recurrent AF (including those with concomitant HF), it is acceptable to allow patients to remain in AF, while being chronically treated not only with AV nodal blocking drugs to achieve adequate ventricular rate control but also with appropriate antithrombotic therapy to prevent thromboembolic complications. ![]() The important question with this rate-control approach is: What defines “adequate” ventricular rate control? While adequate ventricular rate control was previously considered to be achieving a heart rate <80 beats/min at rest and <100 beats/min during exercise, evidence from the RACE II trial has suggested that selecting a more lenient rate-control strategy (resting heart rate <110 beats/min) may be a reasonable approach for certain patients with AF.31 In this trial, a lenient rate-control strategy (resting heart rate <110 beats/min) was considered to be noninferior to a strict heart rate-control strategy (resting heart rate <80 beats/min and heart rate during moderate exercise <110 beats/min) with regard to the primary end point of cardiovascular death, hospitalization for HF, stroke, systemic embolism, bleeding, and life-threatening arrhythmic events. According to the most recent treatment guidelines for AF, this lenient rate-control strategy is recommended for those patients with persistent AF who have no or acceptable symptoms and stable LV function (LVEF >40%).22 In patients with LV systolic dysfunction (LVEF ≤40%), a stricter rate-control approach (resting heart rate <80 beats/min) should be considered to minimize the potential harmful effects of a rapid heart rate response on ventricular function.
The important question with this rate-control approach is: What defines “adequate” ventricular rate control? While adequate ventricular rate control was previously considered to be achieving a heart rate <80 beats/min at rest and <100 beats/min during exercise, evidence from the RACE II trial has suggested that selecting a more lenient rate-control strategy (resting heart rate <110 beats/min) may be a reasonable approach for certain patients with AF.31 In this trial, a lenient rate-control strategy (resting heart rate <110 beats/min) was considered to be noninferior to a strict heart rate-control strategy (resting heart rate <80 beats/min and heart rate during moderate exercise <110 beats/min) with regard to the primary end point of cardiovascular death, hospitalization for HF, stroke, systemic embolism, bleeding, and life-threatening arrhythmic events. According to the most recent treatment guidelines for AF, this lenient rate-control strategy is recommended for those patients with persistent AF who have no or acceptable symptoms and stable LV function (LVEF >40%).22 In patients with LV systolic dysfunction (LVEF ≤40%), a stricter rate-control approach (resting heart rate <80 beats/min) should be considered to minimize the potential harmful effects of a rapid heart rate response on ventricular function.
As in the acute setting, the selection of an AV nodal blocking drug to control ventricular rate in the chronic setting should be primarily based on the patient’s LV function.16 In patients with normal LV function (LVEF >40%), an oral β-blocker, diltiazem, or verapamil is preferred over digoxin because of their relatively quick onset and maintained efficacy during exercise. When adequate ventricular rate control cannot be achieved with one of these drugs, the addition of digoxin may result in an additive lowering of the heart rate. Verapamil and diltiazem should not be used in patients with LV dysfunction (LVEF ≤40%). Instead, β-blockers (i.e., metoprolol succinate, carvedilol, or bisoprolol) and digoxin are preferred in these patients, as these drugs are also concomitantly used to treat chronic HF. Specifically, in patients with NYHA class II or III HF, β-blockers should be considered over digoxin because of their survival benefits in patients with LV systolic dysfunction. If patients are having an episode of decompensated HF (NYHA class IV), digoxin is preferred as first-line therapy to achieve ventricular rate control because of the potential for worsening HF symptoms with the initiation and subsequent titration of β-blocker therapy. If adequate ventricular rate control during rest and exercise cannot be achieved with β-blockers, non-DHP CCBs, and/or digoxin in patients with normal or depressed LV function, oral amiodarone can be used as alternative therapy to control the heart rate.16
Because a rate-control strategy is now considered a reasonable initial approach for the chronic management of AF, the question that remains to be answered is, “In which patients should restoration of SR be considered?” Electrical or pharmacologic cardioversion should be considered for those patients with AF who remain symptomatic despite having adequate ventricular rate control or for those patients in whom adequate ventricular rate control cannot be achieved. In addition, a rhythm-control strategy may be considered in patients who are experiencing their first episode of AF if they are likely to convert to and remain in SR. Chronic AAD therapy is usually not needed in the latter population since the AF is often self-limiting.
In those patients in whom it is decided to restore SR, one must consider that this very act (regardless of whether an electrical or pharmacologic method is chosen) places the patient at risk for a thromboembolic event. The reason for this heightened risk is that the return of SR restores effective contraction in the atria, which may dislodge poorly adherent thrombi. Administering antithrombotic therapy prior to cardioversion not only prevents clot growth and the formation of new thrombi but also allows existing thrombi to become organized and well adherent to the atrial wall. It is a generally accepted principle that the risk of thrombus formation and a subsequent embolic event increases if the duration of the AF exceeds 48 hours. Therefore, it is vital for clinicians to estimate the duration of the patient’s AF, so that appropriate antithrombotic therapy can be administered prior to cardioversion if needed.
According to the most recent guidelines on antithrombotic and thrombolytic therapy developed by the American College of Chest Physicians (ACCP), patients with AF lasting at least 48 hours or an unknown duration should be given therapeutic anticoagulation with warfarin (INR target range 2 to 3), a low-molecular-weight heparin (subcutaneously at treatment doses), or dabigatran for 3 weeks before cardioversion.20 If the 3 weeks of therapeutic warfarin, low-molecular-weight heparin, or dabigatran therapy is not feasible, patients may alternatively undergo transesophageal echocardiography (TEE) to provide guidance regarding the need for antithrombotic therapy prior to cardioversion. If no thrombus is noted on TEE, these patients can be cardioverted without the mandatory 3 weeks of warfarin, low-molecular-weight heparin, or dabigatran pretreatment. In these patients, anticoagulation therapy with either IV unfractionated heparin or a low-molecular-weight heparin (subcutaneously at treatment doses) should be administered during the TEE and cardioversion procedure to prevent the formation of thrombi during the pericardioversion and postcardioversion periods. Cardioversion should then be performed within 24 hours of the TEE. Alternatively, warfarin therapy (INR target range of 2 to 3) may be used for at least 5 days prior to the TEE and cardioversion. If cardioversion is successful, therapeutic anticoagulation with either warfarin (INR target range of 2 to 3) or dabigatran should be continued for at least 4 weeks, regardless of the patient’s baseline risk of stroke. The reason for continuing anticoagulation for this additional 4-week time period is that after restoration of SR, full atrial contraction does not occur immediately. Rather, it returns gradually to a maximum contractile force over a 3- to 4-week period. Decisions regarding long-term antithrombotic therapy after this 4-week time period should be primarily based on the patient’s risk for stroke and not on whether he or she is in SR. If a thrombus is seen on TEE, cardioversion should not be performed and the patient should be anticoagulated indefinitely. If cardioversion is considered in these patients at a later time, a TEE should again be performed. Overall, the use of a TEE-guided approach to cardioversion in patients with AF has been compared with the conventional 3 weeks of anticoagulation before cardioversion in a large, multicenter, randomized trial.32 In this trial, the incidence of thromboembolic events was not different between the two strategies, but bleeding episodes were higher in the group that received 3 weeks of warfarin therapy before cardioversion. Patients in the TEE strategy group had a higher success rate of achieving SR, probably because it is more difficult to terminate AF the longer a patient remains in this arrhythmia.
In patients with AF that is less than 48 hours in duration, anticoagulation prior to cardioversion is unnecessary because there has not been sufficient time to form atrial thrombi.20 However, it is recommended that these patients should receive either IV unfractionated heparin or a low-molecular-weight heparin (subcutaneously at treatment doses) at presentation prior to and when proceeding to cardioversion. If these patients have risk factors for stroke, a TEE could alternatively be performed prior to cardioversion to exclude the presence of thrombus. If cardioversion is successful, therapeutic anticoagulation with either warfarin (INR target range 2 to 3) or dabigatran should be continued for at least 4 weeks, regardless of the patient’s baseline risk of stroke. Decisions regarding long-term antithrombotic therapy after this 4-week time period should be primarily based on the patient’s risk for stroke and not on whether he or she is in SR.
After prior anticoagulation or TEE, the process of restoring SR can be considered. There are two methods of restoring SR in patients with AF or atrial flutter: pharmacologic cardioversion and DCC. The decision to use either of these methods is generally a matter of clinical preference. The disadvantages of pharmacologic cardioversion are the risk of significant side effects (e.g., drug-induced TdP),33 the potential for drug–drug interactions (e.g., digoxin–amiodarone), and the lower efficacy of AADs when compared with DCC. The advantages of DCC are that it is quick and more often successful (80% to 90% success rate). The disadvantages of DCC are the need for prior sedation/anesthesia and a risk (albeit small) of serious complications such as sinus arrest or ventricular arrhythmias.
Nonetheless, despite the relatively high success rate associated with DCC, clinicians often elect to use AADs first, and then resort to DCC in the event that these drugs fail. Pharmacologic cardioversion appears to be most effective when initiated within 7 days after the onset of AF.16 According to the most recent treatment guidelines for AF, there is relatively strong evidence for efficacy of the class III pure IK blockers (ibutilide and dofetilide), the class Ic AADs (e.g., flecainide and propafenone), and amiodarone (oral or IV) within this time frame.16 Class Ia AADs have limited efficacy or have not been adequately studied in this setting. Sotalol is not effective for cardioversion of paroxysmal or persistent AF. Single, oral loading doses of propafenone (600 mg) and flecainide (300 mg) are effective compared with placebo for conversion of recent-onset AF and have been incorporated into the “pill-in-the-pocket” approach endorsed by the treatment guidelines.16,34 With this method, outpatient, patient-controlled self-administration of a single, oral loading dose of either flecainide or propafenone can be a relatively safe and effective approach for the termination of recent-onset AF in a selected patient population that does not have sinus or AV node dysfunction, bundle-branch block, QT interval prolongation, Brugada syndrome, or SHD.35 In addition, this treatment regimen should only be considered in patients who have previously been successfully cardioverted with these drugs on an inpatient basis. In patients with AF that is longer than 7 days in duration, only dofetilide, amiodarone, and ibutilide have proven efficacy for cardioversion.16 The class Ia and Ic AADs have limited efficacy or have been inadequately studied in this setting.
Overall, when considering pharmacologic cardioversion, the selection of an AAD should be based on whether the patient has SHD (e.g., LV dysfunction, CAD, valvular heart disease, LV hypertrophy).16 In the absence of any type of SHD, the use of a single, oral loading dose of flecainide or propafenone is a reasonable approach for cardioversion. Ibutilide can also be used as an alternative in this patient population; however, use of this agent is restricted to a monitored setting in the hospital because it requires QT interval monitoring. In patients with underlying SHD, flecainide, propafenone, and ibutilide should be avoided because of the increased risk of proarrhythmia; amiodarone or dofetilide should be used instead. Although amiodarone can be administered safely on an outpatient basis because of its low proarrhythmic potential, dofetilide therapy can only be initiated in the hospital (for QT interval monitoring). Additionally, it should be remembered that a patient’s ventricular rate should be adequately controlled with AV nodal blocking drugs prior to administering a class Ic (or Ia) AAD for cardioversion. The class Ia and Ic AADs may paradoxically increase ventricular response. Traditionally, this observation has been attributed to the vagolytic action of these drugs despite the fact that only disopyramide displays significant anticholinergic side effects. Therefore, a more likely alternative explanation exists: all of these drugs slow atrial conduction, decreasing the number of impulses reaching the AV node; as a result, the AV node paradoxically allows more impulses to gain entrance to the ventricular conduction system, thereby increasing ventricular rate.
Long-Term Complications There are two forms of therapy that the clinician must consider in each patient with AF: long-term antithrombotic therapy to prevent stroke and long-term AADs to prevent recurrences of AF. Consider the issue of antithrombotic therapy first. Historically, warfarin has been the standard of care for stroke prevention in patients considered to be moderate or high risk for stroke. However, while warfarin is undoubtedly effective in preventing strokes in patients with AF, its use can be associated with a number of potential limitations, including a narrow therapeutic window, requirement for INR monitoring, food and drug interactions, and pharmacogenetic influences. Therefore, researchers have long been searching for an antithrombotic therapy that could be used as an alternative or even as a replacement for warfarin in patients with AF. Over the past few years, several oral antithrombotic therapies have been approved by the Food and Drug Administration for stroke prevention in patients with AF. These oral anticoagulant drugs include the direct thrombin inhibitor, dabigatran, and the factor Xa inhibitors, rivaroxaban and apixaban.
When initiating chronic antithrombotic therapy in patients with AF, assessing the patient’s risk for stroke becomes important for selecting the most appropriate regimen. Based on the most recent ACCP guidelines, the CHADS2 index continues to be recommended for stroke risk stratification in patients with AF.20 With this risk index, patients with AF are given two points if they have a history of a previous stroke or transient ischemic attack, and one point each for being ≥75 years old, having hypertension, having diabetes, or having congestive HF (CHADS2 is an acronym for each of these risk factors). The points are added up and the total score is then used to determine the most appropriate antithrombotic therapy for the patient (Fig. 8-6). Patients with a CHADS2 score of ≥2, 1, or 0 are considered to be at high risk, intermediate risk, and low risk for stroke, respectively. Based on the most recent ACCP guidelines, oral anticoagulant therapy is preferred over aspirin or aspirin plus clopidogrel therapy in patients who are at either high or intermediate risk for stroke. With regard to selection of an oral anticoagulant in both high-risk and intermediate-risk patients with AF, the updated ACCP guidelines suggest that dabigatran should be used rather than warfarin (INR target range 2 to 3). Either no antithrombotic therapy or aspirin is recommended for patients who are at low risk for stroke; however, no therapy is preferred in these patients. If the decision is made to initiate antithrombotic therapy in these low-risk patients, aspirin (75 to 325 mg/day) can be used.
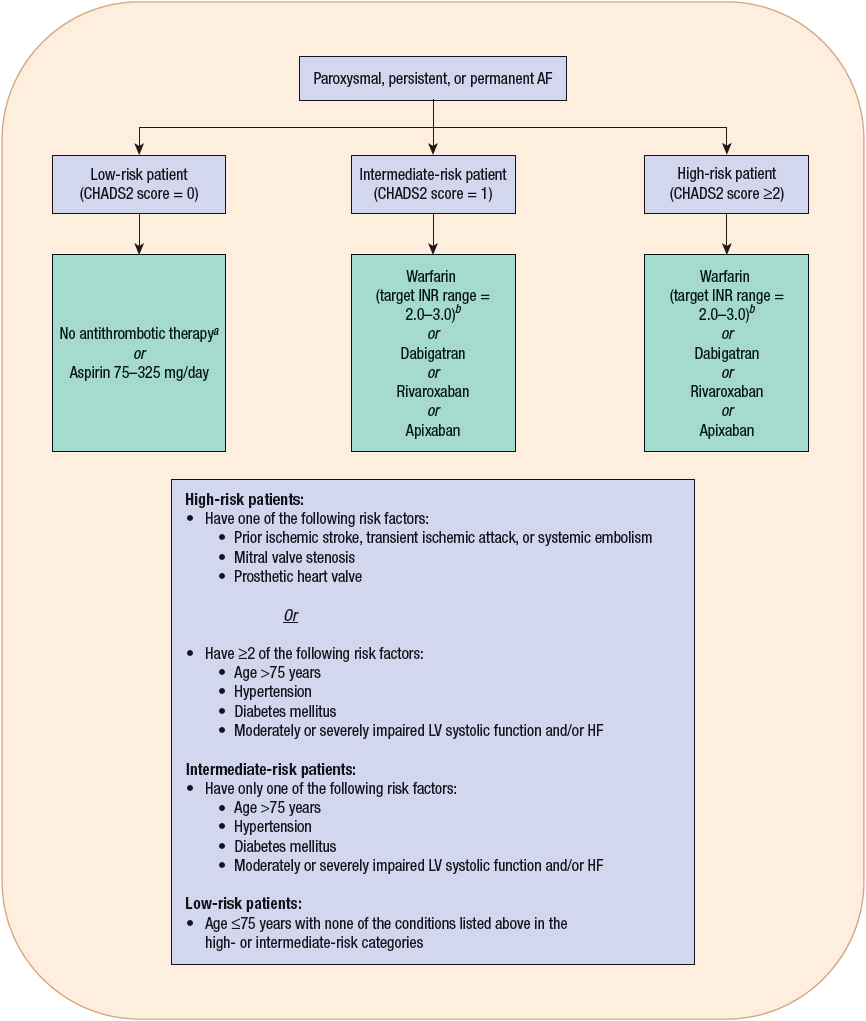
FIGURE 8-6 Algorithm for the prevention of thromboembolism in paroxysmal, persistent, or permanent AF. aNo antithrombotic therapy preferred for low-risk patients. bThe target INR for patients with prosthetic heart valves should be based on the type of valve that is present. (AF, atrial fibrillation; HF, heart failure; INR, international normalized ratio; LV, left ventricular.)



