FIGURE 1 | The main steps of translation.
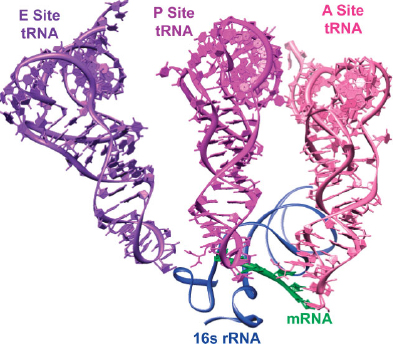
FIGURE 2 | Crystal structure of three tRNA molecules interacting with an mRNA molecule and the 16s rRNA in the 30S ribosome (PDB: 1GIX, 5.50 Å).
Although other RNA sequences have been explored as potential drug targets, the bacterial ribosome remains the only validated one with clinically approved drugs.4,5 Multiple fundamental and practical reasons account for this. The ribosome encompasses a large portion of the cellular RNA and contains distinct and well-defined binding sites for small molecules, which is not necessarily common with less structured cellular RNA targets.6 Small molecules with moderate affinity may exhibit practical potency due to the sheer volume of rRNA in the cell and the intricate function of its components. From a medicinal chemistry perspective, the bacterial ribosome is an attractive target, since its RNA components are often coded by multiple operons, circumstances that may slow down the evolution of resistance.
The fundamental forces dictating RNA–ligand interactions have been discussed elsewhere.7,8 We note that the major groove of RNA duplexes, associated with a large negative electrostatic potential, can be rendered more accessible to small molecules by bulges and loops. This further exposes the hydrogen bonding face and the aromatic surfaces of the nucleobases for additional intermolecular interactions with potential binders.9 The minor groove of RNA duplexes frequently accommodates ligands through hydrophobic interactions. On a more global level, diverse models have been offered to explain the dynamic and structural characteristics of RNA–ligand recognition.10,11,12,13,14,15,16 Conformational capture assumes RNA, being structurally dynamic, populates a variety of conformational states, which may or may not be related to the ultimate biological function.10,11,12 Ligand binding locks a specific conformation. Alternatively (although the boundaries between these models are clearly vaguely defined), ligand binding may induce structural transformations, a process classically referred to as induced fit.17,18 The binding of highly cationic ligands to RNA has been described by a spatial electrostatic complementarity model, where the drugs’ covalently-linked three-dimensional array of positively charged groups complements in space the negative electrostatic potential created by the RNA fold.13,14,15,16 Frequently, cations natively bound in the highly negative potential cavities of RNA can be displaced by positively charged ligands.15,16
Prior to the mid-1990s, the binding sites of antimicrobial agents were elucidated by ribosomal mutations that conferred resistance, as well as cross-linking and footprinting experiments.19 Modern biophysical methods, such as NMR and X-ray crystallography, dramatically enhanced our knowledge of the structure, function, and dynamics of the ribosome. Structural information at the atomic level now provides a window into the actual interactions between antibiotics and the targeted ribosomal constituents.20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43 These contemporary techniques and the data they produce are being used for a structure-based design of new candidate antibacterials.44 Although computational methods for structure-based drug design have shown promise, their application for addressing RNA targets remains limited. In particular, scoring functions for molecular docking have been primarily developed for ligand–protein complexes, which do not always translate well for describing ligand–RNA interactions.14 The flexibility of the phosphate backbone, hydration shell, and divalent ions, which are all important for RNA–ligand interactions, represent challenging parameters to capture in RNA-friendly computational programs.45,46 It is also important to note that, while crystal structures can significantly aid in small molecule drug design, the ribosome is dynamic, occupying different conformations throughout translation. This has been illustrated by the differences in the crystal structures of ribosomes in the apo form and when bound to tRNA mimics.47 Targeting highly dynamic domains with small molecules is likely to remain challenging.
Antibiotics that target the ribosome almost exclusively bind to one of the three key sites: the decoding (or A-site) on the 30S, the PTC on the 50S, and the peptide exit tunnel on the 50S. Antibiotics that bind the A-site, such as the aminoglycosides, interfere with codon recognition and translocation. Peptide bond formation is inhibited when small-molecules like oxazolidinones bind at the PTC. Finally, macrolides tend to block the growth of the amino acid chain at the peptide exit tunnel. Notably, these antibiotics almost solely interact with the RNA components of the bacterial ribosome. We therefore classify the antibacterial agents discussed below according to their cognate ribosomal targets. We note, however, that this crude categorization is put in place for organizational purposes only, as the specific sites as well as the mode of binding and actual molecular contacts of individual antibiotics, even when grouped together, may vary.
THE RIBOSOMAL DECODING SITE
Codon–anticodon recognition and discrimination, performed at the A-site located on the 30S subunit, are critical for translation. Crystal structures of the 30S ribosomal subunit and the whole ribosome of Thermus thermophilus with mRNA and cognate tRNAs show the universally conserved bases A1492, A1493, and G503 interacting with the codon–anticodon minihelix,22,24 which was originally proposed based on NMR structures of the A-site48 (Figure 3(a) and (b), Note Escherichia coli’s nomenclature is applied throughout). As the cognate tRNA enters the A-site, conformational changes occur in the rRNA to initiate peptide bond formation. A1492 and A1493 flip from a syn to an anti conformation and interact in the minor groove of the first two base pairs in the anticodon–codon helix to scrutinize hydrogen bonding patterns. G503 also flips into an anti conformation and interacts with the second anticodon and the third codon base pairs (Figure 3(a) and (b)). It tolerates non-Watson–Crick hydrogen base-pairs, consistent with the degeneracy in the genetic code. Lowered stability of the codon–anticodon minihelix effectively assists in the rejection of noncognate tRNA. This efficient decoding process occurs with extremely low error rates (3 × 10−3 for E. coli).49
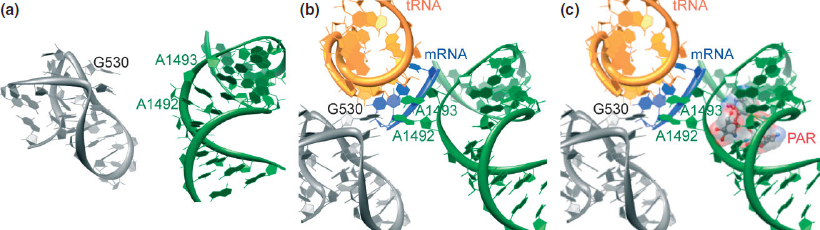
FIGURE 3 | (a) Crystal structure of a section of the 30S ribosomal subunit (PDB: 1J5E, 3.05 Å). (b) Crystal structure of a portion of the 30S ribosomal subunit complexed with an mRNA fragment and cognate tRNA anticodon stem-loop bound at the A-site (PDB: 1IBM, 3.31 Å). (c) Crystal structure of a portion of the 30S ribosomal subunit complexed with an mRNA fragment and cognate tRNA anticodon stem-loop bound at the A-site with paromomycin (PDB: 1IBL, 3.11 Å).
As originally probed by biochemical and footprinting techniques, the bacterial rRNA decoding site is the natural target of most aminoglycosides.50 The majority of these natural products, isolated from Streptomyces, contain the highly conserved 2-deoxystreptamine (2-DOS) aminocyclitol, which is glycosylated with aminosugars at the 4-, 4,6-, or 4,5-positions (Figure 4). The amine groups on the aminoglycosidic scaffold possess a range of pKa values, but they are mostly protonated at physiological pHs.51,52 Thus, electrostatic and hydrogen bonding interactions contribute to their RNA binding.53 While aminoglycoside antibiotics have evolved to bind the bacterial decoding site and meddle with protein biosynthesis, their highly cationic character and conformational flexibility facilitates binding to diverse RNA targets.15 In this respect, they can be viewed as rather universal or somewhat promiscuous RNA binders.15,16
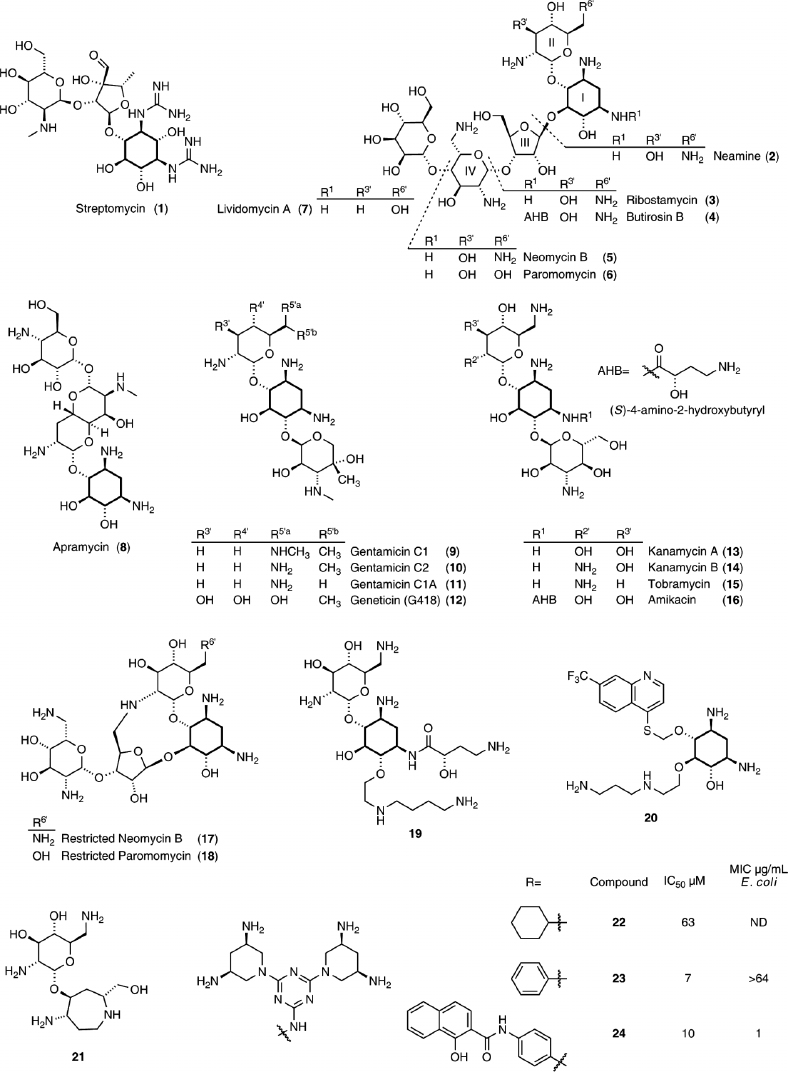
The A-site is an autonomous RNA domain capable of mimicking the function and aminoglycosides recognition features of the entire 16S rRNA.54,55 This has paved the way for the use of short RNA constructs for both biochemical as well as NMR and X-ray crystallography studies.56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71 Additionally, structures with various aminoglycosides bound to the 30S ribosomal subunit and the entire bacterial ribosome were solved.20,21,22,23,24,38,41 Although there are inherent differences in resolution and scope among the solved structures, they provide a consistent molecular picture on the overall interactions of aminoglycosides. It is now established that aminoglycoside antibiotics bind the decoding site and stabilize an RNA conformation similar to the one induced by the cognate acyl-tRNA–mRNA. Nucleobases A1492, A1493, and G530 flip outside the helix to an anti position (Figure 3(c)). This disrupts the decoding process, lowers its fidelity and ultimately increases the misincorporation of amino acids72,73,74; however, evidence also suggests that aminoglycosides interfere with translocation.75,76,77,78,79,80 It is worth noting that streptomycin (1), the only guanidinium-containing aminoglycoside as well as the first to be isolated and clinically used, displays a distinct binding site and mode of action; it interferes with initial tRNA selection.21
Crystal structures have revealed that the paromamine/neamine/gentamicin cores exhibit highly conserved interactions in paromomycin (6),21,62 neomycin B (5), kanamycin A (13), gentamicin C1A (11), ribostamycin (3), lividomycin A (7),65 and amikacin (16).68 Positions N3 and N1 hydrogen bond to N7(G1494) and O4(U1495), respectively. Additionally, N3 forms a salt bridge with O1P(A1493) and O2P(G1494). Ring II stacks against G1491, O5′ bonds with N6(A1408), and the 6′ functional group, a hydroxyl in paromomycin and an amine in the others listed, interacts with N1(A1408) (Figure 5(a) and (b)). These observations suggest that rings I and II represent the key pharmacophore of the aminoglycosides. The 2-DOS ring is now viewed as a privileged RNA binding scaffold.81,82
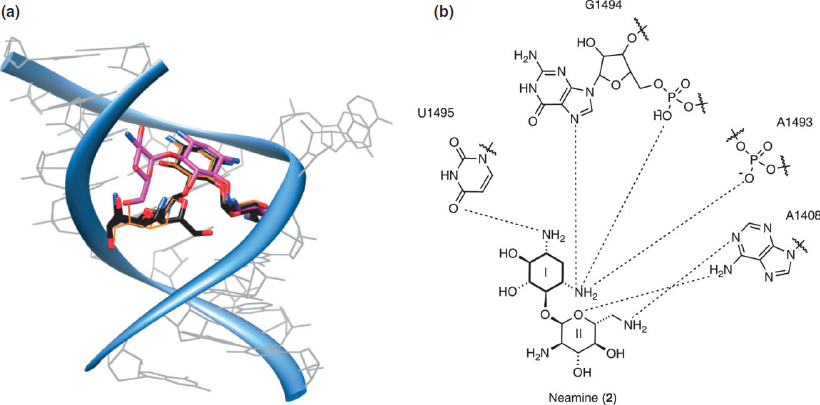
FIGURE 5 | (a) An overlap of the crystal structures of neomycin (PDB: 2ET4, 2.40 Å), tobramycin (PDB: 1LC4, 2.54 Å), and paromomycin (PDB: 1J7T, 2.50 Å) in an A-site model construct. (b) Conserved contacts of neamine.
Aminoglycosides typically display specificity toward prokaryotes over eukaryotes. This is facilitated by both differences in membrane permeability and intrinsic target specificity.83,84,85,86 In contrast to prokaryotic ribosomes, which contain an adenosine at 1408 and a guanine at 1491, eukaryotic rRNA contains a guanine and an adenine, respectively, at these positions, which are essential for aminoglycoside recognition.65 Point mutations in bacteria, such as A1408G, lowers sensitivity to many 2-DOS containing aminoglycosides, and when these bacteria contain only one copy of rRNA operon, total resistance ensues.84,87 Importantly, human mitochondrial ribosomes that have A1408 and G1491 at analogous positions exhibit higher resemblance to their bacterial counterparts. This similarity is likely responsible for some of the adverse effects shown by aminoglycosides (e.g., ototoxicity).83,88,89
Not unlike many other antibiotics, the clinical use of aminoglycosides has been compromised by the emergence of resistant bacteria. The prominent resistant mechanisms rely on enzymatic modifications of the antibiotics, including O-adenylation, O-phosphorylation, and N-acetylation90; all lower their affinity to the rRNA target. The innate flexibility of the glycosidic bonds in aminoglycosides promotes their binding to resistance enzymes,91 which likely evolved to kill competitor and predator organisms and to provide self immunity to the producing organism.92,93,94 Through horizontal gene transfer, this self-preserving trait could have passed onto other bacteria.93,94 Many other resistant mechanisms exist, as well, such as the operation of efflux pumps, rRNA modifications, and mutations in ribosomal proteins.95 The removal of aminoglycosides through efflux pumps was first reported in Burkholderia pseudomallei and has been observed in many nosocomial infections, such as Pseudomonas aeruginosa.96,97 Modification of rRNA include the methylation of N1 in A1408 and N7 in G1405, and it results in resistance to some 4,6-substituted 2-DOS aminoglycosides.92,98
To combat the rise in bacterial resistance, while improving the efficacy and circumventing the toxicity of known drugs, new aminoglycoside derivatives have been sought.99,100,101 Early as well as more recent efforts involved diverse modifications of the natural products.102,103,104,105,106 For example, amikacin (Figure 4, 16), containing an acylated 2-DOS ring, is a semisynthetic aminoglycoside that is highly effective against bacterial inactivating enzymes. The presence of the L-hydroxyaminobutyramide residue (AHB) at N1 lowers its susceptibility toward acylation and phosphorylation.95,107 More recent approaches have been rather diverse and include conformational restriction of aminoglycosides,67,108,109 the functionalization of 2-DOS with carbohydrate free moieties,110,111,112,113,114,115 replacement of the 2-DOS core with mimetic scaffolds,116 or creation of totally synthetic molecules.117
As previously mentioned, aminoglycosides exhibit some degree of flexibility around their glycosidic bonds, allowing them to conformationally adapt and bind diverse targets.15 To explore specificity, conformationally restricted neomycin (17) and paromomycin (18, Figure 4) derivatives were synthesized to study A-site binding and antibacterial activity.67,108 The crystal structure of the conformationally locked neomycin derivative (17) bound to the A-site exhibited the identical intermolecular interactions between rings I and II as neomycin (5).67 The affinity and antibacterial activity decreased, however, when compared to the natural product. This has been attributed to the impact this intramolecular cyclization had on basicity of the amine at position 2′.118 Intriguingly, this modification has also been examined in the context of antibiotic resistance and the susceptibility to resistance enzymes of the restricted neomycin derivative (17) was evaluated.109 Crystal structures of certain aminoglycosides bound to resistance enzymes exhibit significantly different binding modes than the bound A-site conformation, inspiring the design for the restricted neomycin analog (17).82 The restriction of the derivative prevents adenylation by Staphylococcus aureus ANT(4′) and Mycobacterium tuberculosis AAC(2′), and accordingly retain antibacterial activity in resistant strains containing these enzymes.109
To maintain conserved contacts, attempts have been made at derivatizing neamine (2), the key aminogycosidic pharmacophore. On the basis of molecular modeling, Mobashery and coworkers prepared neamine analogs with AHB on N1, similar to amikacin, and aminoaliphatic chains on O6.112 The crystal structure of the A-site bound derivative 19 shows that both arms provide additional contacts to RNA. Superimposition of this compound with the neamine core of kanamycin bound to the phosphotransferase resistance enzyme, APH(3′)-IIIa, suggested that the AHB substitution hinders binding.119,120
Aminoglycoside mimetics containing noncarbohydrate moieties in place of the natural sugars rings (e.g., aminoalkyl chains) have been synthesized.110,111,112 Another approach reported by Griffey and coworkers involved replacing Ring II on aminoglycosides cores with aromatic groups.113,114 The two approaches were combined to create carbohydrate free aminoglycoside mimetics containing 2-DOS, with compound 20 showing inhibition of bacterial translation.115
As 2-DOS appears to be a privileged RNA binding scaffold,4 DOS-mimetics can, in principle, maintain essential A-site contacts. Hermann and coworkers replaced 2-DOS in neamine with azepane glycosides. Compound 21 showed moderate activity against S. aureus and other aminoglycoside resistant strains.116 Another set of derivatives, based on 3,5-diamimino-piperidinyl triazines (DAPT) as 2-DOS mimetics, was shown to inhibit protein synthesis, but had weak antibacterial activity. Further structural refinement generated derivatives 22–24 that demonstrated increased potency against some Gram-negative bacteria.117
To facilitate the discovery of A-site targeting agents and new antibacterials, numerous approaches have been explored.121,122 In addition to the use of mass spectrometry and NMR techniques, simpler biophysical assays have been developed.123,124,125,126,127 Fluorescent A-site constructs, which contain 2-aminopurine at positions 1492 or 1493, have shown great promise, as they rely on changes in the location and dynamics of these residues.128,129 A robust analysis platform for antibiotics targeting the bacterial A-site has also been developed by incorporating a new emissive uridine surrogate into the RNA and labeling the aminoglycosides with a coumarin. Detection of antibiotic binding and displacement is dependent on the interaction between the two chromophores acting as a förster resonance energy transfer (FRET) pair.130 This approach has recently been further advanced to facilitate the rapid determination of the prokaryotic versus eukaryotic selectivity of A-site binders.131
ANTIBIOTICS TARGETING THE PEPTIDYL TRANSFERASE CENTER
The PTC connects all functional cores in the ribosome, including the tRNA entrance and exit regions, as well as the A-site. In essence, it is the ribosomal active site, catalyzing peptide bond formation by meticulously positioning the reactive partners.132,133,134 Not surprisingly, numerous antibiotics target this key site, including chloramphenicol (25), clindamycin (26), tiamulin (27), sparsomycin (28), and the streptogramins (29–32), as well as the oxazolidinones (33–35), the only fully synthetic class of antibiotics (Figure 6). These antibiotics either hinder tRNA substrate binding or disrupt peptide bond formation by binding to the PTC itself.
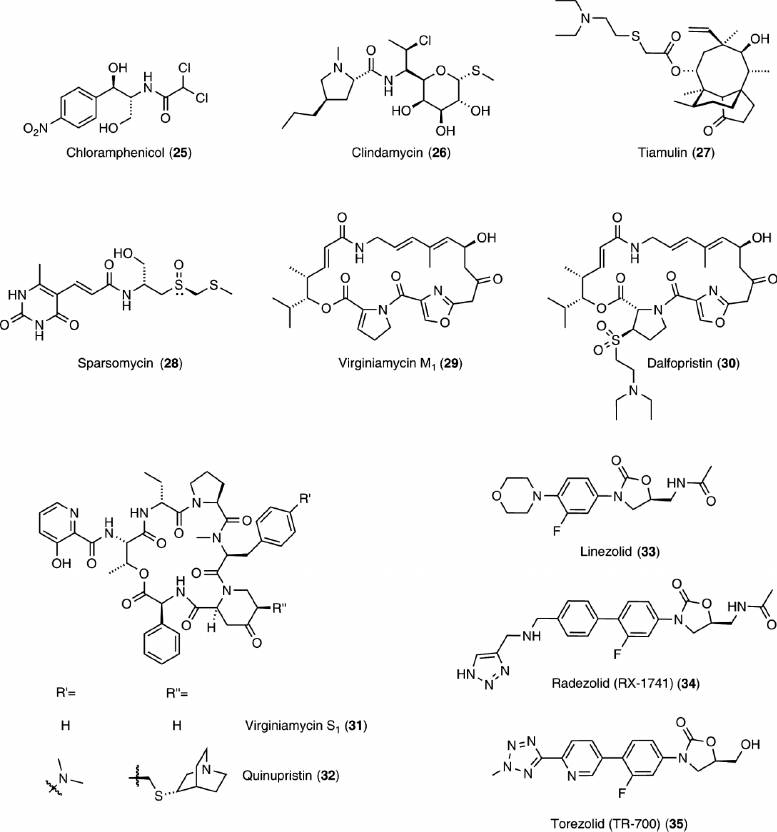
Chloramphenicol (25), one of the first naturally occurring antibiotics to be produced synthetically, interferes with A-site and tRNA binding, preventing peptide bond formation.26,29,135,136 On the other hand, clindamycin (26) and tiamulin (27) interfere with both A-site and P-site tRNA substrate binding. Overlapping the chloramphenicol (25) binding site, clindamycin (26), a lincosamide, binds at the PTC and peptide exit tunnel.27,29 It inhibits the peptidyl transferase reaction and causes dissociation of peptidyl tRNA from the ribosome.137,138 Tiamulin (27), a member of the pleuromutilin class of antibiotics, inhibits peptide bond formation.139,140,141 It competes with chloramphenicol (25), clindamycin (26), and the saccharide branch of carbomycin A (41) for binding.25,29,36,141,142
Sparsomycin (28), a uracil derivative, is an effective protein synthesis inhibitor of both bacterial and eukaryotic ribosomes.143,144 It has been shown to be necessary for a tRNA to be bound in the P-site for sparsomycin to bind,145 and crystal structures with the Haloarcula marismortui 50S subunit (H50S) suggest extensive interactions between sparsomycin and the CCA 3′-end of the P-site tRNA substrate analog.26 It can bind, however, to Deinococcus radiodurans 50S subunit (D50S) alone and causes a great conformational change in the PTC, which could be the origin of its inhibitory activity on peptide bond formation.33 Although sparsomycin competes with chloramphenicol (25), it does not competitively inhibit A-site tRNA binding.143
The streptogramin A antibiotics (29 and 30), a family of unsaturated macrocyclic depsipeptides (Figure 6), typically bind at the PTC and overlap the A-site and P-site tRNA, likely interfering with tRNA binding.26,27,35 In contrast, the streptogramin B antibiotics (31 and 32), a family of macrocyclic depsipeptides (Figure 6), bind in the peptide exit tunnel in the macrolide binding region,27,35 possibly impeding peptide elongation. Notably, while individual members of each family show modest bacteriostatic activity, the two families act synergistically and display greater bactericidal effects.146,147,148
Importantly, the PTC is targeted by the oxazolidinones, a family of synthetic derivatives representing one of the very few success stories of antibiotic development. Exploring these derivatives as orally available antimicrobial agents started in the 1970s, ultimately leading to the introduction of linezolid (33) into the clinic a decade ago. It is active against many gram-positive bacteria, including methicillin resistant S. aureus (MRSA) and vancomycin-resistant enterococci (VRE).149,150
Early studies suggested linezolid (33, Figure 6) bound to the 30S and 50S subunits,151 but newer cross linking studies have indicated binding only at the PTC.152,153 Crystal structures of linezolid (33) bound to H50S with and without CCA-N-acetylphenylalanine (CCA-Phe) show a drug molecule in the PTC and a CCA-Phe molecule in the P-site.28 It appears likely that oxazolidinones compete with the A-site substrate, binding near the bases A2451, U2585, and U2506 (Figure 7(a)). These bases are shown to be important in A-site-tRNA binding and the positioning of the substrates for the peptidyl transferase reaction.134 Although alternative structures show slightly distinct contacts, the general location of linezolid (33) bound at the PTC is similar (Figure 7(c)).28,42
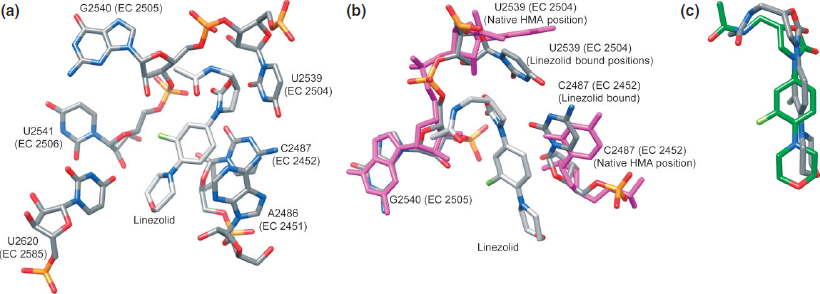
FIGURE 7 | (a) Crystal structure of linezolid bound to the H50S (PDB: 3CPW, 2.70 Å). (b) Crystal structure of linezolid bound to the H50S (PDB: 3CPW, 2.70 Å) overlapped with the crystal structure of native H50S (PDB: 3CC2, 2.40 Å). (c) Crystal structure of linezolid bound to the H50S (PDB: 3CPW, 2.70 Å) overlapped with the crystal structure of linezolid bound to the D50S (PDB: 3DLL, 3.50 Å).
The binding specificity of linezolid (33) may be explained by comparing the 50S subunit of the bacteria E. coli (E50S) and archaeon H. marismortui.28 In H50S, when linezolid (33) is bound, the conformation of U2504 is nearly identical to the homologous base in E50S with no ligand24 (Figure 7(b)), indicating that the binding pocket for the drug is prearranged in bacterial ribosomes and more rigid. Oxazolidinones have been shown, however, to bind to and possibly inhibit archaeal, bacterial, and mitochondrial ribosomes, but not those from human cytoplasm.153,154 It is also suggested that the position of U2504 in eukaryotic ribosomes could possibly be incompatible with linezolid (33) binding.42 In addition, cross-resistance between PTC binders occurs due to their overlapping binding sites near U2504.37 Almost all nucleotides conferring resistance are clustered on one side of the PTC near nucleotide U2504. About half of the nucleotides mediating resistance do not directly interact with bound PTC antibiotics, but reshape the binding pocket, often by affecting the conformation and/or flexibility of U2504.37
Bacterial resistance to linezolid (33) is currently minimal,1 but as for all other antibacterials, it is expected to escalate. The only mechanism of clinical resistance recorded so far is the target modification of the PTC, with a G2576U mutation found in Enterococci and S. aureus.155 Although resistance is currently uncommon for linezolid (33), as recognition by efflux pumps and resistance enzymes of a completely synthetic molecule should not exist, search is underway for novel oxazolidinones. One example worth mentioning is hybrid molecules inspired by the partial overlap seen in the binding sites of linezolid (33) and sparsomycin (28).26,28,156,157 Radezolid (RX-1741) (34) and Torezolid (TR-700) (35) represent two derivatives that are in advanced clinical trials (Figure 6).158,159
PEPTIDE EXIT TUNNEL AS A TARGET
Toward the end of protein synthesis, the newly synthesized peptide chain travels through a dynamic tunnel below the PTC, lined with mostly rRNA, and emerges approximately 100 Å out of the 50S subunit. While the exit tunnel is primarily straight, there is a bend 20–35 Å away from the PTC. In this bent section, L22 and L4, two ribosomal proteins, constrain the tunnel into its narrowest part.160 At this region, the peptide sequence can influence gating, or pause elongation and termination.161,162,163 Antibiotics, such as the macrolides, which bind this domain, impede the progression of the nascent peptides.
Macrolides, a family of naturally occurring 12- to 18-membered macrocyclic lactones, typically contain one or more deoxy monosaccharides (Figure 8). Erythromycin (36) is a prototypical example, as it was the first to be clinically used. Semisynthetic derivatives of erythromycin (36) include 14- and 15-membered ring macrocycles (37–40), as well as ketolides (44–46). Macrolides and ketolides bind to the peptide exit tunnel and prevent elongation,25,27,29,31,32,34 consequently inhibiting protein synthesis.138 This results in premature dissociation of short peptidyl–tRNAs from the ribosome,138 which was originally misinterpreted as inhibition of peptidyl transferase or translocation.164 The length of the resulting short terminated peptides depends on the individual antibiotic.138,165,166
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


