- Tetracyclines are broad-spectrum agents with excellent bioavailability and activity against both Gram positive and Gram negative bacteria; resistance and issues of toxicity restrict their regular clinical use.
- New classes of tetracycline are being developed; the first clinical example, tigecycline, a glycylcycline, shows potency across a wide range of bacteria and low susceptibility to resistance.
- For further information, see Chopra and Roberts (2001) and Zhanel et al. (2004).
Tetracycline antibiotics used clinically include doxycycline, minocycline, and tetracycline, while tigecycline, which has the same four-ringed structure and is a derivative of minocycline, is the first member of the glycylcyclines to be approved; their structures are shown in Figure 4.3.1 and their therapeutic indications are listed in Table 4.3.1.
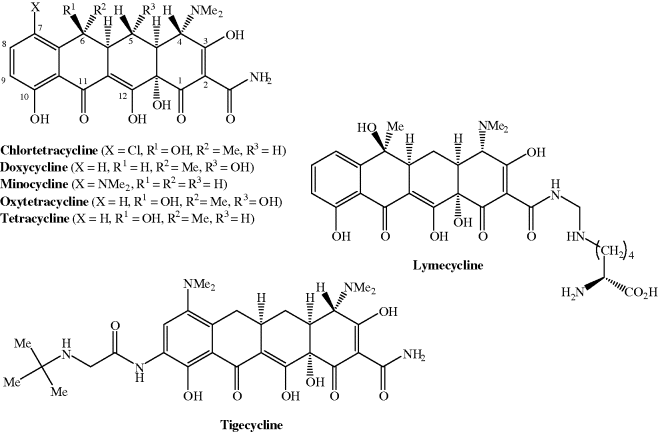
Table 4.3.1 Therapeutic indications for the tetracycline antibiotics.
| Tetracycline antibiotic | Indications |
| Doxycycline | Chronic prostatitis, sinusitis, syphilis, uncomplicated genital chlamydial infection, pelvic inflammatory disease, acne vulgaris, rosacea, Lyme disease, community-acquired pneumonia |
| Lymecycline | Acne vulgaris |
| Minocycline | Acne vulgaris, prophylaxis of asymptomatic meningococcal carrier state (no longer recommended) |
| Oxytetracycline | Acne vulgaris, rosacea |
| Tetracycline | Acne vulgaris, rosacea, non-gonococcal urethritis, chronic bronchitis |
| Tigecycline | Complicated intra-abdominal or skin/soft tissue infections |
4.3.1 Discovery
The tetracycline antibiotics are the third example so far in this section, of naturally occurring molecules from a microbial source that interfere with bacterial protein synthesis. Once again, the first in this series, chlortetracycline (originally named aureomycin), resulted from a programme of screening soil microorganisms for potential new antibiotics. This discovery is attributed to Benjamin M. Duggar, a retired botanist, with expertise across a wide range of plant and microorganism physiology. He retired from Wisconsin University in 1943, when he was 71, but was approached to act as consultant for Lederle Laboratories in New York (part of American Cyanamid Company, now part of Wyeth Pharmaceuticals), who were supporting the war effort by searching for new antibiotic and antimalarial agents (Walker, 1982).
Although he is solely credited with the discovery of chlortetracycline in 1948, Duggar was part of a larger team, under the direction of Yellapragada SubbaRow, which was systematically investigating soil microorganisms for natural products with desirable pharmaceutical activities. Duggar requested some local samples from the soil microbiologist at the University of Missouri, William Albrecht, from which he cultured a golden mould, which produced a yellow pigment that displayed growth inhibitory properties against bacteria, such as streptococci. He identified the mould as a Streptomyces species that had not previously been catalogued; to reflect its colour, he named it Streptomyces aureofaciens and the antibiotic it produced aureomycin (Duggar, 1948). Note that the ‘mycin’ part of the name corresponds with its isolation from a Streptomyces species, like the aminoglycoside streptomycin and the macrolide erythromycin, which we have just met.
Aureomycin was quickly released to clinicians and other researchers to obtain evaluative data of its activity and efficacy; it gathered support with glowing testimonials of its broad spectrum of activity, including against streptomycin- and penicillin-resistant organisms (Wright and Schreiber, 1949; Cantor, 1950; Kiser et al., 1952). It was found to be as effective as penicillin and streptomycin and had the significant advantage of being the first antibiotic that was effective when administered orally.
Following the discovery of aureomycin, other tetracyclines were soon discovered:
- Oxytetracycline (originally called terramycin) in 1949 from Streptomyces rimosus by Pfizer (Finlay et al., 1950).
- Tetracycline (marketed originally as achromycin (Darken et al., 1960)) in 1953 from S. aureofaciens when cultured with a chlorination inhibitor (Goodman and Matrishin, 1968).
- Demethylchlortetracycline in 1957 from S. aureofaciens (McCormick et al., 1957; Wilson, 1961). This was the last natural tetracycline to be identified and was originally called declomycin or ledermycin. It is still marketed as the latter (or under its generic name, demeclocycline) by Lederle Laboratories.
The discovery of this new class of antibiotics is not without controversy, though: Pfizer, American Cyanamid, and Bristol-Myers formed a monopoly that maintained artificially high prices for tetracycline over several years before the US Federal Trade Commission halted the violations after a series of high-profile investigations, charges, and appeals heard in the high court (Anon, 1964; US Court of Appeals, 1968).
When it was discovered that the hydrogenation of chlortetracycline resulted in dechlorination and conversion into tetracycline (which was as active as chlortetracycline) (Stephens et al., 1952; Conover, 1955), the possibility that synthetic modification of tetracyclines might provide alternative agents with antibacterial activity was realised. During the next 15–20 years, many semi-synthetic analogues were prepared, including lymecycline, doxycycline, and minocycline; some of these second-generation semi-synthetic tetracyclines were even more potent than chlortetracycline and are still marketed today. Structural modification has continued and has resulted in the discovery of a third-generation tetracycline, t-butylglycylamidominocycline (tigecycline, originally labelled GAR-936) (Petersen et al., 1999), with more in development and in clinical trials (Sun et al., 2008; Brötz-Oesterhelt and Sass, 2010).
Structurally, the tetracyclines are based on a four-ring (tetracyclic or octahydronaphthacene) system, hence the name; the rings are labelled A, B, C, and D (Figure 4.3.2). One face consists of carbonyl, phenol, alcohol, and enol oxygen atoms, with high polarity and metal ion binding ability, while the other face is substantially less polar. There are a number of substitution patterns commonly found in the antibiotic tetracyclines and significant deviation from these leads to greatly reduced antibacterial activity (Chopra and Roberts, 2001; Zhanel et al., 2004).
Figure 4.3.2 Requirements for tetracycline antibiotic activity (Chopra and Roberts, 2001; Zhanel et al., 2004)
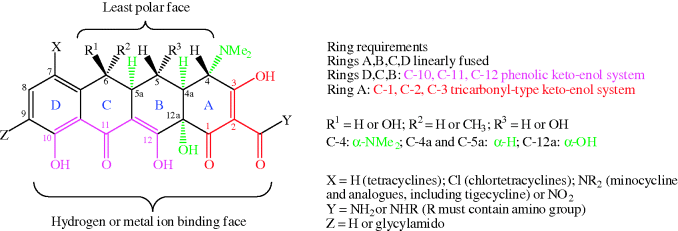
Much of the development of new tetracycline antibiotics has been driven by the instability of the first-generation tetracyclines, particularly chlortetracycline, oxytetracycline, and tetracycline, which can lead to degradation during storage and even production of a toxic product. We will look at some of the reactions of tetracyclines and the resultant effects upon bioavailability in Subsection 4.3.3.
4.3.2 Synthesis
Looking back at Figure 4.3.1, you can see that the tetracyclines have a number of chiral centres and functional groups, so you will not be surprised to learn that fermentation methods are considered to be the most cost-effective for their production, and for the production of the base structures for semi-synthetic analogues, such as lymecycline and tigecycline (Khosla and Tang, 2005). As you will see further on in this subsection, there is now an efficient chemical synthetic method for multigram quantities of a key intermediate in tetracycline synthesis, which offers the possibility of analogue synthesis (Brubaker and Myers, 2007). The first patented fermentations of S. aureofaciens were for the production of chlortetracycline (Duggar, 1948; Neidercorn, 1952) and tetracycline (Goodman et al., 1959); much work since then has focussed on optimising the selectivity for, and the yields of, the desired tetracyclines, especially since some Streptomyces species can produce more than one tetracycline, depending upon the fermentation conditions (Bêhal, 1987, 2000). In early 2011, there were almost 3000 patents relating to the biosynthesis and synthesis of tetracycline and its analogues (worldwide.espacenet.com), including some filed in 2010 and 2011 – proof that there is still interest in the production and use of tetracyclines. It should be noted, however, that some of these were for non-antibiotic uses of tetracyclines, briefly mentioned in Subsection 4.3.4.
4.3.2.1 Biosynthesis of Tetracyclines
The biosynthesis of tetracycline antibiotics is related to the bacterial synthesis of fatty acids through the bacterial type II polyketide synthase pathway, consisting of a well-studied set of enzymes (for examples, see Khosla, 2009; Zhang and Tang, 2009), although the synthesis of tetracyclines is unique to certain bacteria (Clardy et al., 2009). The biosynthetic pathways to tetracycline and oxytetracycline are the most studied (for example, Petkovi et al., 2006; Pickens and Tang, 2009). The biosynthetic pathway to natural tetracyclines is available from the KEGG database (http://www.genome.jp/kegg/pathway/map/map00253.html, last accessed 10 March 2012); in summary, it uses the precursor, malonamyl coenzyme A (CoA), which is obtained from acetyl CoA via malonyl CoA and glutamine (Wang et al., 1986), and proceeds through two common key intermediates, 6-methylpretetramide and 4-ketoanhydrotetracycline (Scheme 4.3.1 and Table 4.3.2) (Clardy et al., 2009; Pickens and Tang, 2009).
et al., 2006; Pickens and Tang, 2009). The biosynthetic pathway to natural tetracyclines is available from the KEGG database (http://www.genome.jp/kegg/pathway/map/map00253.html, last accessed 10 March 2012); in summary, it uses the precursor, malonamyl coenzyme A (CoA), which is obtained from acetyl CoA via malonyl CoA and glutamine (Wang et al., 1986), and proceeds through two common key intermediates, 6-methylpretetramide and 4-ketoanhydrotetracycline (Scheme 4.3.1 and Table 4.3.2) (Clardy et al., 2009; Pickens and Tang, 2009).
Scheme 4.3.1 Biosynthetic pathway to tetracycline antibiotics (Clardy et al., 2009; http://www.genome.jp/kegg/pathway/map/map00253.html, last accessed 10 March 2012)
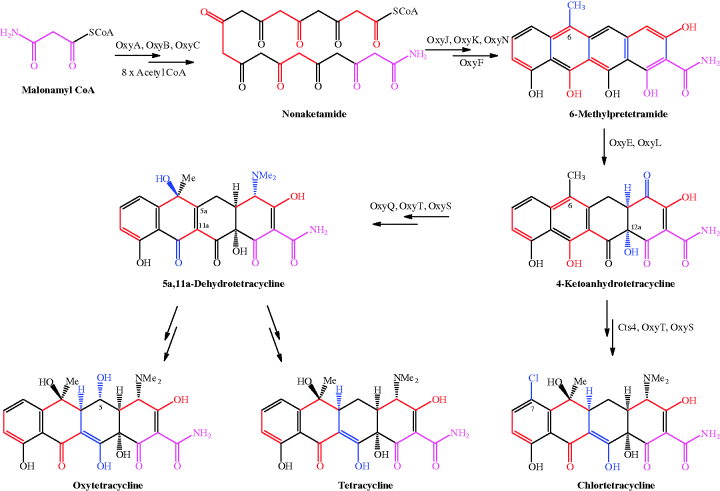
Table 4.3.2 Enzymes involved in tetracycline biosynthesis.
| Enzyme | Function |
| OxyA | Ketosynthase |
| OxyB | Chain length factor |
| OxyC | Acyl carrier protein |
| OxyJ | Ketoreductase |
| OxyK | Aromatase |
| OxyN | Cyclase |
| OxyF | C-methyltransferase |
| OxyE | Flavin-dependent monoxygenase |
| OxyL | NADPH-dependent dioxygenase |
| OxyQ | Aminotransferase |
| OxyT | N,N-dimethyltransferase |
| OxyS | Monooxygenase that hydroxylates stereospecifically at C6 |
| Cts4 | Halogenase |
For clarity, in Scheme 4.3.1 the precursor unit, malonamyl CoA, is coloured pink throughout; the consecutive acetyl units added to malonamyl CoA from acetyl CoA are coloured alternately black and red – you can see that the cycle of acetyl addition occurs eight times until the linear nonaketamide is formed. A series of enzymic reactions involving OxyJ, OxyK, and OxyN leads to pretetramide (not shown in Scheme 4.3.1), which is converted into 6-methylpretetramide by OxyF. This series of reactions results in cyclisation of the nonaketamide to the tetracyclic structure of 6-methylpretetramide, followed by methylation at C6, with the new bonds formed in this sequence shown in blue. If you trace the sequentially added acetyl units, you can see how the two-carbon units form the backbone of the structure. Oxidation at C4 (by OxyE), and hydroxylation at C12a by OxyL, provides the second key intermediate, 4-ketoanhydrotetracycline, at which the tetracycline biosynthetic paths diverge. Chlortetracycline results from Cts4 halogenase action at C7 (Dairi et al., 1995), followed by amination at C4 (OxyQ) and OxyT N,N-dimethylation (the methyl groups are provided by S-adenosyl methionine); OxyS catalyses the stereospecific hydroxylation at C6, leaving a stereospecific reduction at C5a required to produce chlortetracycline. In the other pathway, amination at C4, dimethylation, and hydroxylation at C6 provide 5a,11a-dehydrotetracycline, from which oxytetracycline and tetracycline are obtained.
Much detailed research has been directed at elucidating this pathway; most was carried out on the enzymes of the oxytetracycline-producing species Streptomyces rimosus, hence the Oxy names, but the enzymes are the same or similar for the other tetracycline-producing species (Zhang et al., 2007; Petkovi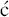 et al., 2010; Pickens and Tang, 2010).
et al., 2010; Pickens and Tang, 2010).
4.3.2.2 Chemical Synthesis of Tetracyclines
The literature related to the chemical synthesis of tetracyclines resembles a ‘Who’s Who’ of synthetic organic chemistry:
- R. B. Woodward solved the structure, complete with stereochemistry, in 1952 (this was revised slightly in the 1960s with the help of X-ray crystallography) (Hochstein et al., 1953; Donohoe et al., 1963; von Wittenau et al., 1965).
- Woodward and Conover (who first synthesised tetracycline by hydrogenation of chlortetracycline) synthesised a biologically active tetracycline, named sancycline (Korst et al., 1968), albeit in 25 steps and 0.002% overall yield.
- Shemyakin synthesised a tetracycline natural product, (±)-12a-deoxy-5a,6-anhydrotetracycline (Gurevich et al., 1967).
- Muxfeldt identified the major problems with the synthesis of tetracyclines: the complexity of the required stereochemistry and the sensitivity of the tetracycline functional groups to both mild acid and base during initial studies (Muxfeldt and Rogalski, 1965), then later achieved the total synthesis of (±)-5-oxytetracycline in 22 steps and 0.06% (Muxfeldt et al., 1968, 1979).
- Stork concentrated on achieving the correct stereochemistry at each centre in the basic tetracycline structure, producing (±)-12a-deoxytetracycline in 16 steps and an impressive 18–25% yield, although this structure has little antimicrobial activity (Stork et al., 1996).
- Tatsuta and co-workers exploited the natural stereochemical definition of carbohydrates for their starting materials and achieved the total synthesis of natural (−)-tetracycline from D-glucosamine in 34 steps and 0.002% yield (Tatsuta et al., 2000; Tatsuka and Hosokawa, 2005), including a solution to the difficult stereospecific hydroxylation of C12a.
- More recently, Myers and co-workers developed a highly effective synthetic approach to natural tetracyclines, their analogues, and their precursors (Charest et al., 2005a, 2005b; Brubaker and Myers, 2007; Myers et al., 2007, 2011; Sun et al., 2008), which takes account of the considerable challenge in achieving the correct stereochemistry, particularly at C12a, and provides versatility for the synthesis of many new analogues for microbiological evaluation (Myers et al., 2007; Sun et al., 2008).
A discussion of the chemical strategies for synthesising tetracyclines, the reactions required, and their stereochemical complexities would be a section in itself, so we will restrict ourselves here to consideration of the most recent syntheses, which have enabled multigram quantities of optically pure tetracyclines to be achieved and thus offer potential commercial synthetic routes to these agents. Myers and colleagues recognised that one key intermediate 1, providing the A and B rings of tetracyclines, allows the synthesis of a wide range of tetracycline antibiotics and their analogues (Scheme 4.3.2).
Scheme 4.3.2 Key intermediates in the synthesis of tetracycline antibiotics (Myers et al., 2007, 2011)
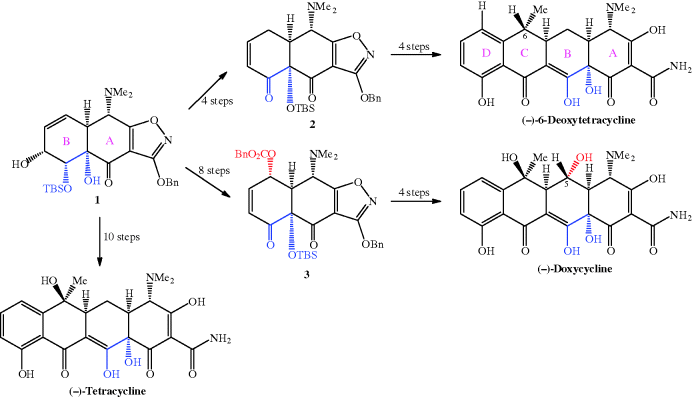
Initially, they developed a synthesis to this important intermediate from benzoic acid and achieved intermediate 1 in 21% overall yield after 7 steps (Charest et al., 2005a). Using this route, (−)-tetracycline could be synthesised in 17 steps and 1.1% yield from benzoic acid. Conversion of 1 into 2 and 3 provided routes to tetracyclines with no hydroxyl group at C5 (including an alternative synthesis of tetracycline) and 5-hydroxytetracyclines, respectively. Using this approach, the synthesis of (−)-6-deoxytetracycline was achieved in 14 steps and 7% yield via intermediate 2, while (−)-doxycycline was isolated in 8.3% yield after 18 steps via intermediate 3; the yields and number of steps relate to the total synthesis from benzoic acid (Charest et al., 2005a, 2005b; Myers et al., 2007, 2011). Not content with these impressive achievements, Myers and Brubaker re-designed and improved the synthesis of intermediate 2 (Scheme 4.3.3), from which most tetracyclines can be accessed; they identified an alternative cheap and readily available commercial starting material, methyl 3-hydroxy-5-isoxazolecarboxylate 4, along with an improved synthetic strategy to improve stereoselectivity and yields, and obtained intermediate 2 in 9 steps and 21% overall yield from starting material 4 (Brubaker and Myers, 2007).
Scheme 4.3.3 Improved synthesis of key intermediate 2 for large-scale tetracycline synthesis (Brubaker and Myers, 2007)
There are several noteworthy features in this revised synthesis of intermediate 2 (Scheme 4.3.3):
- First, the introduction of a stereogenic (chiral) centre into structure 5 is carried out in high yield and with a high enantiomeric excess, and this enantiomeric ratio is maintained during the SN2 replacement of the hydroxyl group (as a mesylate) with a dimethylamino group in the synthesis of 6.
- The resulting stereogenic centre at C6 becomes C4 in the tetracycline and already has the correct stereochemistry, which is retained throughout the remainder of the synthesis.
- Intermediate 7 has a new stereogenic centre bearing a hydroxyl group; you may be concerned that the stereochemistry is not defined at this new centre, but this group is oxidised to a carbonyl during the synthetic sequence that results in intermediate 8, so the mixed stereochemistry does not matter at this stage.
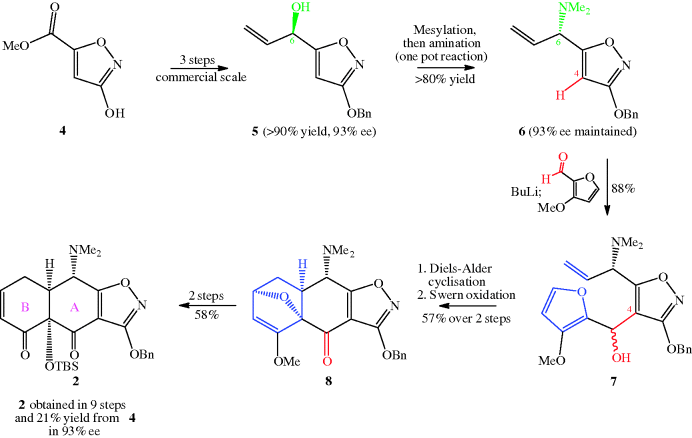
So far, we have not considered how rings C and D can be constructed, yet this is just as important for the tetracycline structure. The synthetic strategy adopted by the Myers group uses intermediate 1, 2, or 3 as appropriate to provide rings A and B of the tetracycline with the correct stereochemistry and functionality, then elaborates this basic structure by construction of the C-ring, while adding the D-ring through a generalised Michael–Dieckmann reaction sequence (using a carbanion formed from a variety of D-ring precursors), followed by deprotection of all the functional groups (Scheme 4.3.4) (Charest et al., 2005a).
Scheme 4.3.4 Elaboration of intermediates 2 and 3 into tetracyclines and their analogues (Charest et al., 2005a)
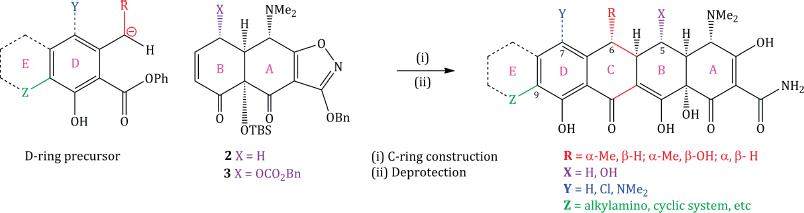
One great advantage of this approach is the ease with which varying functionality can be added throughout the structure, particularly substituents at C5 (X), C6 (R), C7 (Y), C9 (Z), and even an extra (E) ring; a great many analogues have been made, and some of these combine strong antibacterial activity with activity against strains resistant to first- and second-generation tetracycline antibiotics (Myers et al., 2011; Sun et al., 2011). The synthetic routes to the tetracyclines developed by the Myers group have been sufficiently successful to support a spin-out company, Tetraphase, which has several tetracyclines in early clinical trials. Other groups have also pursued C9-substituted tetracyclines (for example, Koza and Nsiah, 2002; Sum et al., 2006); the clinical success of tigecycline (see Subsections 4.3.4 and 4.3.5) and the development of amadacycline (which is in clinical trials and is discussed in Subsection 4.3.9) provided the rationale for their evaluation.
4.3.3 Bioavailability
The pharmacokinetics and pharmacodynamics of the tetracycline antibiotics were reviewed recently (Agwuh and MacGowan, 2006; Barbour et al., 2010), but they are not fully understood, with several seemingly contradictory observations. The tetracyclines display time-dependent effects, yet the general concentration-dependent parameters (of exposure time at a concentration above the MIC) provide good clinical results, with a strong post-antibiotic effect. Although generally considered to be bacteriostatic, there is evidence of bactericidal activity with certain tetracycline antibiotics against specific bacteria, when used at an appropriate concentration (Zhanel et al., 2004; Barbour et al., 2010).
The instability of the first-generation tetracyclines during storage has already been mentioned; we will consider the reactions of tetracyclines more carefully here, as they have an effect upon bioavailability and even upon the safety of the products.
The first-generation tetracyclines, although clinically successful, were found to be unstable to acidic, basic, and neutral pH during storage and in solution, including in the gastrointestinal (GI) tract after administration, decreasing their bioavailability (Walton et al., 1970; Ali and Strittmatter, 1978; Wu and Fassihi, 2005). Two main reactions occur in the presence of acid: epimerisation at C4, to produce epitetracycline (Hussar et al., 1968) (Scheme 4.3.5), and dehydration of 6-hydroxytetracyclines across C5a-C6 (with loss of the OH group at C6), to give the anhydrotetracycline derivative, as demonstrated for tetracycline in Scheme 4.3.6.
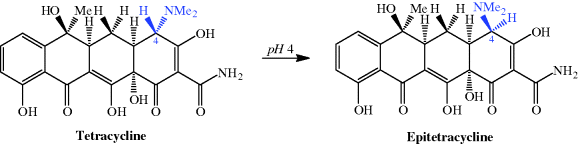
Scheme 4.3.6 Acid-catalysed dehydration of chlortetracycline and improved stability of demethylchlortetracycline (demeclocycline)
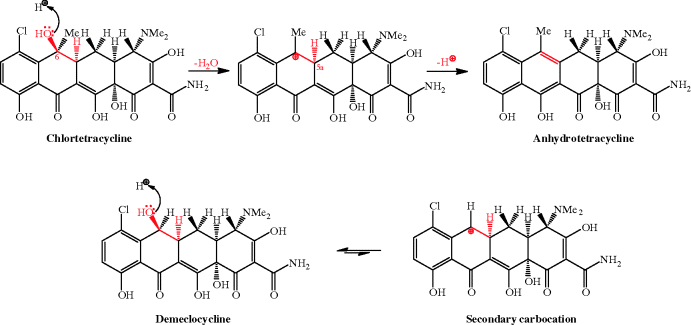
The dehydration (shown in the upper part of Scheme 4.3.6 for chlortetracycline) proceeds through an E1 mechanism, involving protonation of the C6-OH and loss of a good leaving group (H2O), to produce a stabilised tertiary carbocation at C6, followed by loss of a proton from C5a to form the alkene group of anhydrotetracycline. The slightly improved acid stability of demeclocycline and slower dehydration is due to fact that the secondary carbocation that has to be formed at C6 is less stable, and this process thus has a greater activation energy. In this latter case, reversible protonation of the C6-OH favours demeclocycline, instead of proceeding to the carbocation (Scheme 4.3.6).
Epimerisation at C4 of the anhydrotetracycline derivatives can also occur, producing the corresponding epianhydrotetracycline derivative (Sokoloski et al., 1977). Some of the products formed by acid- or base-catalysed degradation are themselves active as antibiotics, while others, for example anhydrotetracycline and epianhydrotetracycline, are toxic (Mull, 1966; Kunin, 1967). The adverse effects (which can manifest as Fanconi syndrome; see Subsection 4.3.7) of using tetracyclines that have degraded during storage were observed soon after these agents had been adopted into regular use, but it was several more years before reliable analytical methods for their analysis and quality control were developed (Frimpter et al., 1963; Gross, 1963; Butterfield et al., 1973). We will discuss the adverse effects of tetracyclines in Subsection 4.3.7; our main concern here is that the bioavailability of some tetracyclines (particularly the first-generation members) can be reduced by their degradation during storage, particularly in solution (Wu and Fassihi, 2005), or as a result of GI-induced reactions (Okeke and Lamikanra, 1995; Dos Santos et al., 1998). The second-generation tetracyclines, doxycycline and minocycline, and the third-generation, tigecycline, are more stable to acidic pH, as they do not have a C6-OH substituent to be protonated and eliminated as water.
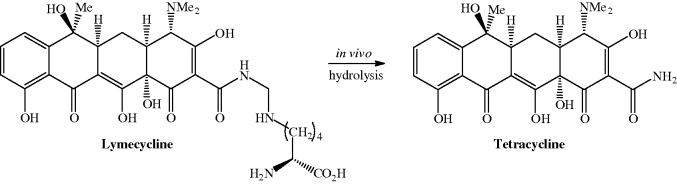
Lymecycline is a prodrug form of tetracycline that is hydrolysed at acidic and neutral pH in vivo to tetracycline, formaldehyde (methanal), and the amino acid lysine after oral and parenteral administration (Scheme 4.3.7). Interestingly, it has lower oral bioavailability than the parent compound, tetracycline (Sjölin-Forsberg and Hermansson, 1984).
The balance of lipophilicity to hydrophilicity in tetracyclines is affected by pH (Chen and Lin, 1998); such a property is usually a clue that there are functional groups in the molecule under investigation which are ionisable at physiological pH values. Each tetracycline has at least one readily ionisable amine group (at C4) and two enol groups (at C3 and C12) that are also relatively easily ionised. First- and second-generation tetracyclines have three pKa values in the physiological pH range (generally around 3.2, 7.6, and 9.6) – the two enol groups, at C3 and C12, are the most acidic (and so have the lower pKa values). The pKa of the phenol group at C10 (~12) means that it is not ionised at physiological pH values. The acid–base equilibria for tetracycline, which are typical of the tetracycline antibiotics, are demonstrated in Scheme 4.3.8 (Jin et al., 2007).
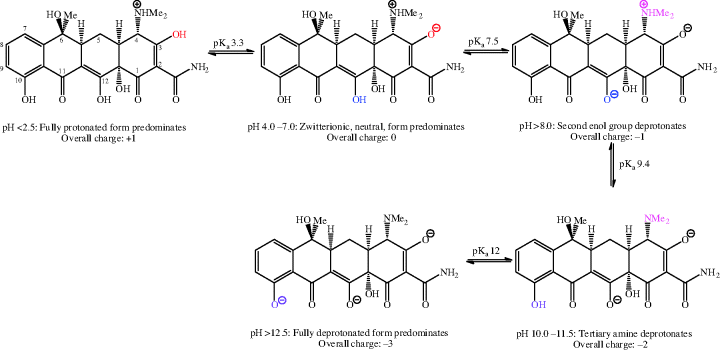
The fully protonated form of the tetracyclines (with an overall charge of +1) is likely to predominate in the acidic medium of the stomach, minimising absorption from this compartment (as neutral species are absorbed best). It is not surprising to find that tetracycline antibiotics are absorbed chiefly from the duodenum, where the pH is around 6–6.5 and the overall neutral form of the tetracycline predominates (Colaizzi and Klink, 1969). By modifying the pH, greater aqueous solubility of the tetracycline antibiotics can be achieved, making them suitable for parenteral administration, and oxytetracycline, lymecycline, doxycycline, and minocycline have been formulated in this way (Chopra and Roberts, 2001).
The third-generation antibiotic tigecycline has limited oral bioavailability and is administered by IV infusion, due to its greater hydrophilicity and reduced lipophilicity as a result of its extra ionisable groups (Meagher et al., 2005). Tigecycline has two extra ionisable groups (Figure 4.3.3):
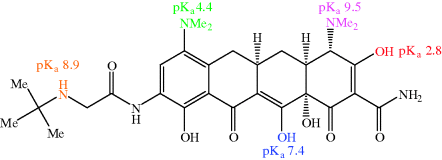
- The t-butylamine on the side chain at C9 (a secondary aliphatic amine, so expected to be basic, with a pKa (of the conjugate acid, R3NH+) of 8.5–10).
- The dimethylamino group at C7 (an aromatic amine, so expected to be a weak base, due to resonance of the nitrogen lone pair with the π-system of the aromatic ring, with a pKa of 3.5–5).
Besides being affected by pH, the absorption of the first-generation tetracyclines in particular, and some of the second-generation analogues, is adversely affected by their concurrent administration with food (Schimdt and Dalhoff, 2002; Agwuh and MacGowan, 2006), dairy products, and other metal-ion-containing preparations (such as antacid treatments), although there is some evidence that the absorption of lymecycline is less affected by milk (Ericson and Gnarpe, 1979). Tetracycline has a logP value of 0.09 and a bioavailability of around 75%, which is reduced by about 50% when co-administered with food (Miller et al., 1977; Zhanel et al., 2004). By comparison, doxycycline and minocycline have a greater bioavailability (90–100%) (Zhanel et al., 2004), their absorption is not affected by food, and they are well absorbed after oral administration. The greater lipophilicity of these agents (logP values of 0.95 and 1.12, respectively) (Colaizzi and Klink, 1969) undoubtedly make a major contribution to these properties. Although minocycline offers improved oral bioavailability over the majority of other tetracyclines, the risk of side effects when used for a protracted period and of the development of resistance has limited it largely to the treatment of acne vulgaris. A recent review suggests that it may offer potential for the systemic treatment of community-associated MRSA and of the significant nosocomial threat of Acinetobacter baumanii (Bishburg and Bishburg, 2009).
Tetracyclines are known to bind strongly to a range of metal ions, with the strongest and most significant binding, in terms of bioavailability, mode of action, mechanisms of resistance, and adverse events, being to magnesium, calcium, iron, and copper (Agwuh and MacGowan, 2006). Chelation of tetracyclines to metal ions in the GI tract adversely affects the absorption of both the tetracycline and the metal ion through the precipitation of insoluble metal-tetracycline complexes (Agwuh and MacGowan, 2006), which provides a scientific rationale for avoiding the administration of tetracyclines alongside metal ion preparations and dairy products. The ability of tetracyclines to coordinate metal ions means that these antibiotics should not be administered to children, due to their ability to sequester calcium and other metal ions at a crucial time in bone and teeth development. In the USA and Australia, children under the age of eight are contraindicated, while in the UK tetracyclines should not be given to children under the age of 12. European guidelines on the third-generation tetracycline tigecycline recommend that it is not used for children and adolescents under the age of 18 years, due to lack of data on its safety and efficacy in these patient groups. The reason for the strong metal ion binding can be seen by considering the tetracycline structure – the lower face of the molecule (as can be seen in Scheme 4.3.9) has several oxygen atoms and so is ideal for binding to a metal ion; the C12 (as the enolate) and C11 oxygen atoms are accepted to be the major binding site (Jin et al., 2007; Palm et al., 2008).
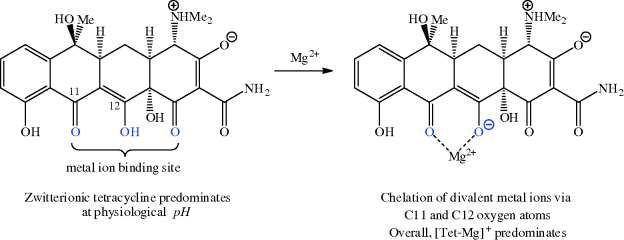
We will return to tetracycline-magnesium complexes again later in this section, when we consider the uptake of tetracyclines into bacterial cells and also when we look at the mode of action (Subsection 4.3.4) and mechanisms of resistance (Subsection 4.3.5).
In general, the tetracycline antibiotics are not metabolised, except for tetracycline, of which about 5% is metabolised, and tigecycline, of which 5–20% is metabolised (Meagher et al., 2005); they are excreted by both the urinary (<50%) and the faecal (>40%) routes (Agwuh and MacGowan, 2006). The urinary excretion of tetracyclines has been found to be affected by pH; as expected, it is significantly increased at pH values above 8, at which values greater ionisation and hydrophilicity are also to be expected (Jaffe et al., 1973).
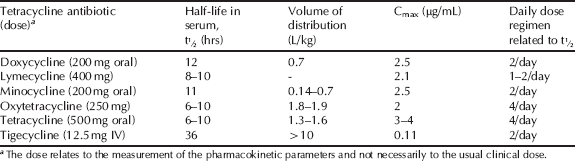
The tetracyclines exhibit a high volume of distribution and generally good tissue penetration; alongside their long half-lives and significant post-antibiotic effect, most can be administered once or twice daily (Table 4.3.3).
Table 4.3.3 Pharmacokinetic parameters for selected tetracycline antibiotics (Zhanel et al., 2004; Agwuh and MacGowan, 2006; Hoffmann et al., 2007; Barbour et al., 2010).
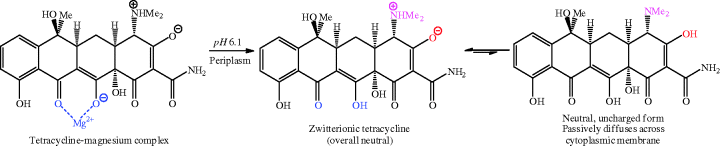
We discussed above how metal ion binding adversely affects the absorption of tetracyclines from the GI tract, but it is an important and crucial part of the uptake of tetracyclines into bacterial cells. The chelation to a magnesium ion forms a tetracycline-magnesium cationic complex (with an overall charge of +1), which is transported through the outer membrane into the periplasm by porins, such as the well-characterised OmpF and OmpC examples from Gram negative bacteria (Nikaido, 1994, 2003). You will remember from Section 1.1.2 that porins are protein pores which transport a range of molecules through the outer membrane into the periplasm; they are known to be responsible for the transport of other antibacterial agents, such as quinolones and β-lactams, besides tetracyclines (Jaffe et al., 1982; Mortimer and Piddock, 1993). The mechanism for porin-mediated transport relies upon the Donnan potential14 across the outer membrane and leads to accumulation of the tetracycline-magnesium cationic complex in the periplasm (Zhanel et al., 2004). It is probable that the tetracycline-magnesium complex dissociates in the periplasm, perhaps due to the lower pH in this compartment, which is sufficiently acidic to drive the reprotonation of the enol oxygen at C12. The released zwitterionic tetracycline (overall neutral charge) is in equilibrium with a small proportion of the uncharged form, which is weakly lipophilic and able to diffuse through the cytoplasmic (inner) membrane in an energy-requiring process (Scheme 4.3.10) (Nikaido and Thanassi, 1993). Tetracyclines are presumed to adopt a similar uncharged tetracycline diffusion entry route through the simpler cell membrane of Gram positive bacteria.
Scheme 4.3.10 A tetracycline-magnesium complex dissociates to the zwitterion in the periplasm and equilibrates with the neutral form, which enters bacterial cells by passive diffusion (Nikaido and Thanassi, 1993)
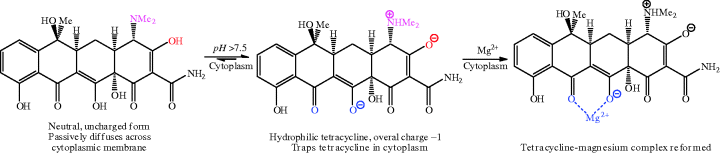
You may be wondering why the diffusion of the neutral tetracycline across the bacterial cytoplasmic membrane is energy-requiring. Live bacteria have a difference in pH between the cytoplasm and periplasm of about 1.7 pH units (Nikaido and Thanassi, 1993), so that, after the neutral tetracycline passes through the cytoplasmic membrane, it becomes trapped at the higher pH of the cytoplasm, releasing a proton to form a greater proportion of tetracycline in a more hydrophilic form (overall molecular charge of −1) (Scheme 4.3.11). To maintain the pH of the cell, and the difference with the external pH, the bacteria must continue to transport protons out of the cytoplasm, which is an energy-dependent process (Nikaido and Thanassi, 1993), so it is not actually the diffusion of tetracycline into the cell that is energy-requiring, but the maintenance of pH that is required as a result.
Scheme 4.3.11 In the cytoplasm, the hydrophilic form of the tetracyclines predominates and reforms the tetracycline-magnesium complex (Nikaido and Thanassi, 1993)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


