T-cell Prolymphocytic Leukemia Involving Lymph Node and Other Tissues
Tariq Muzzafar, MBBS
L. Jeffrey Medeiros, MD
Key Facts
Terminology
T-PLL is aggressive disease characterized by
Numerous small/medium-sized T prolymphocytes
Involvement of blood, bone marrow, spleen, liver, and skin
Clinical Issues
Median age: ˜ 62 years; male predominance
T-PLL is widespread at time of initial diagnosis
Median WBC: 40 × 109/L (range: 1.6-621)
Poor prognosis
Microscopic Pathology
Features of T-PLL in H&E stained tissue sections
Small to medium-sized cells
Nucleoli identified in subset of cells
Oil immersion often needed to appreciate nucleoli
Lymph node
Paracortical or diffuse replacement of architecture
Cytologic features best observed in PB and BM smears
Typical, small cell, and cerebriform variants
Ancillary Tests
Pan-T-cell antigens(+), TCR-αβ(+), CD52(+)
CD4(+), CD8(-) or CD4(+), CD8(+)
Chromosome rearrangements in T-PLL
inv(14)(q11q32) or t(14;14)(q11;q32): ˜ 70% cases
t(X;14)(q28;q11): < 10% cases
Top Differential Diagnoses
Adult T-cell leukemia/lymphoma (ATLL)
Sézary syndrome/mycosis fungoides
Blastic plasmacytoid dendritic cell neoplasm
T-cell large granular lymphocytic leukemia
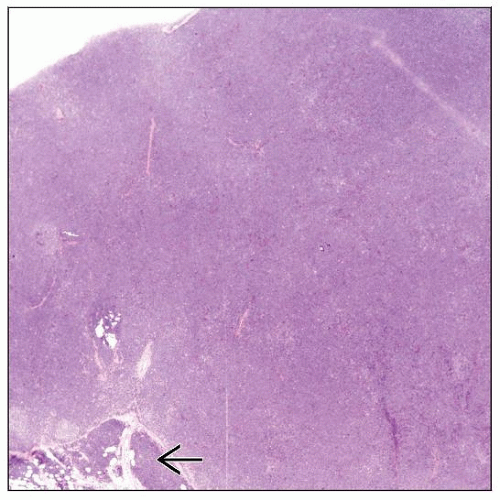 Low-power magnification of a lymph node involved by T-cell prolymphocytic leukemia (T-PLL). The nodal architecture is effaced. The neoplastic cells extend through the capsule into fat  . . |
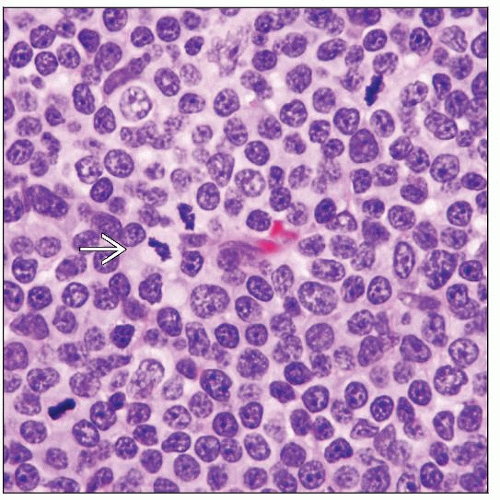 Lymph node involved by T-PLL. The neoplastic cells are small to medium-sized, and nucleoli are not prominent. Four mitotic figures  are present in this field. are present in this field. |
TERMINOLOGY
Abbreviations
T-cell prolymphocytic leukemia (T-PLL)
Synonyms
T-chronic lymphocytic leukemia
Has been used for small cell variant of T-PLL
Definitions
Aggressive leukemia characterized by
High number of small to medium-sized prolymphocytes
Involvement of peripheral blood (PB), bone marrow (BM), spleen, and liver ± other sites
Mature T-cell lineage
ETIOLOGY/PATHOGENESIS
Environmental Exposure
No known role for radiation or carcinogenic agents
Infectious Agents
No known role for any virus
Role of Inheritance
No familial clustering of cases has been reported
Patients with ataxia-telangiectasia (AT) are at increased risk for T-PLL
Ataxia-Telangiectasia Mutated (ATM) Gene Mutations in T-PLL
AT patients have germline mutations in ATM gene at chromosome 11q23
˜ 10% of AT homozygotes develop malignancy, particularly lymphomas or T-PLL
Suggests ATM as candidate tumor suppressor gene in pathogenesis of T-PLL
AT patients can develop T-cell clones without evidence of T-PLL
May be subsequently followed by overt T-PLL
In patients without AT, ATM mutations occur in 60-70% of T-PLL cases
ATM-deficient transgenic mice have increased frequency of T-cell neoplasms
Chromosomal Rearrangements in T-PLL
inv(14)(q11q32) or t(14;14)(q11;q32) result in
TCRα/δ gene at 14q11 juxtaposed with TCL-1 gene at 14q32
Results in T-cell leukemia-1 (TCL-1) gene activation and expression
t(X;14)(q28;q11) results in
TCRα gene juxtaposed with MTCP-1 gene at Xq28
MTCP-1 is homologous with TCL-1
TCL-1 and MTCP-1 in transgenic mouse models induce lymphoma
Long latency suggests that other oncogenic events are required for lymphomagenesis
Tcl-1 Protein Overexpression in T-PLL
Tcl-1 is not expressed by normal mature T cells but is expressed in 70-80% of cases of T-PLL
Tcl-1 dysregulation via chromosomal rearrangement is unique to T-PLL
Normal functions of Tcl-1
Role in normal T-cell physiology is not well understood
Tcl-1 protein product is β barrel protein normally located in cytoplasm
Engagement of T-cell receptor (TCR) leads to recruitment of Tcl-1 and Akt to membrane
Tcl-1 forms activation complexes with Akt and TCR kinases
Tcl-1 binds with Akt and regulates its activity, possibly promoting Akt transphosphorylation
In T-PLL, Tcl-1 may augment TCR responsiveness or perhaps substitute for TCR engagement
Tcl-1 dysregulation may drive early clonal expansion or promote growth advantage
Possibly most important in early stages of pathogenesis of T-PLL
With clonal evolution, other molecular abnormalities may drive proliferation
CLINICAL ISSUES
Epidemiology
Incidence
Rare
˜ 2% of mature lymphocytic leukemias in patients > 30 years
Age
Median: ˜ 62 years
Gender
Male predominance
Male:Female ratio reported up to ˜ 3:1
Ethnicity
No ethnic predisposition or geographical clustering
Site
T-PLL is usually widespread at time of diagnosis
Peripheral blood and bone marrow involved in virtually all patients
Splenomegaly in ˜ 75% patients
Hepatomegaly in ˜ 50%
Lymphadenopathy in ˜ 25-50%
Skin lesions in ˜ 25%
Serous effusions in ˜ 15%; more common at relapse
CNS, conjunctiva, and lung are rarely involved
Presentation
Most patients present with evidence of aggressive disease
Leukocytosis and absolute lymphocytosis in peripheral blood
Rapidly rising leukocyte count
Thrombocytopenia in 45%; anemia in 25% of patients
Due to bone marrow failure &/or hypersplenism
Splenomegaly can be massive
> 10 cm below costal margin in many patients
Can cause local mass-type symptoms or hypersplenism
Lymphadenopathy is usually generalized
Skin involvement is variable
Symmetrical rash with petechial/purpuric features
Facial involvement with swelling
Diffuse infiltrative erythema and nodules may occur
Erythroderma is unusual
Skin rash after established diagnosis is followed by aggressive clinical course
“Smoldering” T-PLL in ˜ 25% of patients
These patients are asymptomatic or relatively well at initial diagnosis
Moderate and relatively stable levels of absolute lymphocytosis in peripheral blood
Patients may have prolonged indolent phase
Median duration: 33 months; rarely can be > 5 years
In almost all patients, disease eventually progresses
Manifested by rapid increase in absolute lymphocyte counts
Demographic features similar to patients in overt aggressive phase
Treatment does not affect duration of indolent phase or risk of progression
Laboratory Tests
Complete blood count
Absolute lymphocytosis with features consistent with prolymphocytes
> 100 × 109L in ˜ 50% of patients
Median in series at MD Anderson Cancer Center (MDACC); Houston, Texas; USA: 40 × 109/L (range: 1.6 – 621)
Thrombocytopenia in 45%; anemia in 25% of patients
Due to bone marrow failure &/or hypersplenism
Treatment
Drugs
Low response rates of short duration using traditional combination chemotherapy regimens
e.g., cyclophosphamide, doxorubicin, vincristine, and prednisolone (CHOP)
2-deoxycoformycin (pentostatin)
Adenosine deaminase inhibitor
Overall response rate (ORR): ˜ 50%
Alemtuzumab (anti-CD52 monoclonal antibody)
As first-line treatment: ORR of 94% and CR rate of 90-100%
Response transient and disease progresses
Given as maintenance therapy and at relapse
Response poor if serous effusions, hepatic or CNS involvement present
May be ineffective if CD52 down-regulated at relapse
Hematopoietic stem cell transplant
If patient achieves response to chemotherapy, used as consolidation
Prolongs disease-free and overall survival
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


