380 | Rheumatoid Arthritis |
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory disease of unknown etiology marked by a symmetric, peripheral polyarthritis. It is the most common form of chronic inflammatory arthritis and often results in joint damage and physical disability. Because it is a systemic disease, RA may result in a variety of extraarticular manifestations, including fatigue, subcutaneous nodules, lung involvement, pericarditis, peripheral neuropathy, vasculitis, and hematologic abnormalities.
Insights gained by a wealth of basic and clinical research over the past two decades have revolutionized the contemporary paradigms for the diagnosis and management of RA. Serum antibodies to cyclic citrullinated peptides (anti-CCPs) are routinely used along with rheumatoid factor as a biomarker of diagnostic and prognostic significance. Advances in imaging modalities have improved our ability to detect joint inflammation and destruction in RA. The science of RA has taken a major leap forward with the identification of new disease-related genes and further deciphering of the molecular pathways of disease pathogenesis. The relative importance of these different mechanisms has been highlighted by the observed benefits of the new class of highly targeted biologic and small-molecule therapies. Despite these gains, incomplete understanding of the initiating pathogenic pathways of RA remains a sizable barrier to its cure and prevention.
The last two decades have witnessed a remarkable improvement in the outcomes of RA. The historic descriptions of crippling arthritis are currently encountered much less frequently. Much of this progress can be traced to the expanded therapeutic armamentarium and the adoption of early treatment intervention. The shift in treatment strategy dictates a new mind-set for primary care practitioners—namely, one that demands early referral of patients with inflammatory arthritis to a rheumatologist for prompt diagnosis and initiation of therapy. Only then will patients achieve their best outcomes.
CLINICAL FEATURES
The incidence of RA increases between 25 and 55 years of age, after which it plateaus until the age of 75 and then decreases. The presenting symptoms of RA typically result from inflammation of the joints, tendons, and bursae. Patients often complain of early morning joint stiffness lasting more than 1 h that eases with physical activity. The earliest involved joints are typically the small joints of the hands and feet. The initial pattern of joint involvement may be monoarticular, oligoarticular (≤4 joints), or polyarticular (>5 joints), usually in a symmetric distribution. Some patients with inflammatory arthritis will present with too few affected joints to be classified as having RA—so-called undifferentiated inflammatory arthritis. Those with an undifferentiated arthritis who are most likely to be diagnosed later with RA have a higher number of tender and swollen joints, test positive for serum rheumatoid factor (RF) or anti-CCP antibodies, and have higher scores for physical disability.
Once the disease process of RA is established, the wrists, metacarpophalangeal (MCP), and proximal interphalangeal (PIP) joints stand out as the most frequently involved joints (Fig. 380-1). Distal interphalangeal (DIP) joint involvement may occur in RA, but it usually is a manifestation of coexistent osteoarthritis. Flexor tendon tenosynovitis is a frequent hallmark of RA and leads to decreased range of motion, reduced grip strength, and “trigger” fingers. Progressive destruction of the joints and soft tissues may lead to chronic, irreversible deformities. Ulnar deviation results from subluxation of the MCP joints, with subluxation of the proximal phalanx to the volar side of the hand. Hyperextension of the PIP joint with flexion of the DIP joint (“swan-neck deformity”), flexion of the PIP joint with hyperextension of the DIP joint (“boutonnière deformity”), and subluxation of the first MCP joint with hyperextension of the first interphalangeal (IP) joint (“Z-line deformity”) also may result from damage to the tendons, joint capsule, and other soft tissues in these small joints. Inflammation about the ulnar styloid and tenosynovitis of the extensor carpi ulnaris may cause subluxation of the distal ulna, resulting in a “piano-key movement” of the ulnar styloid. Although metatarsophalangeal (MTP) joint involvement in the feet is an early feature of disease, chronic inflammation of the ankle and midtarsal regions usually comes later and may lead to pes planovalgus (“flat feet”). Large joints, including the knees and shoulders, are often affected in established disease, although these joints may remain asymptomatic for many years after onset.
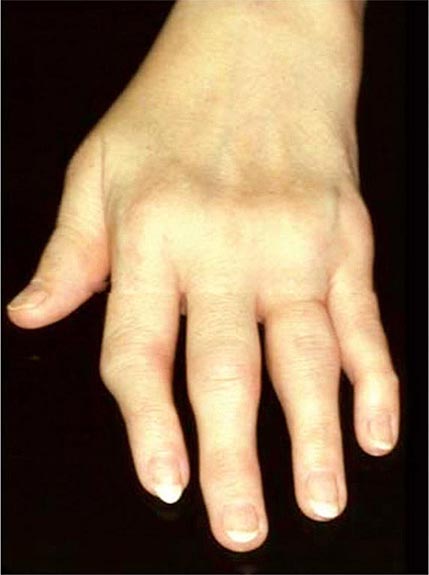
FIGURE 380-1 Metacarpophalangeal and proximal interphalangeal joint swelling in rheumatoid arthritis. (Courtesy of the American College of Rheumatology Image Bank.)
Atlantoaxial involvement of the cervical spine is clinically noteworthy because of its potential to cause compressive myelopathy and neurologic dysfunction. Neurologic manifestations are rarely a presenting sign or symptom of atlantoaxial disease, but they may evolve over time with progressive instability of C1 on C2. The prevalence of atlantoaxial subluxation has been declining in recent years, and occurs now in less than 10% of patients. Unlike the spondyloarthritides (Chap. 384), RA rarely affects the thoracic and lumbar spine. Radiographic abnormalities of the temporomandibular joint occur commonly in patients with RA, but they are generally not associated with significant symptoms or functional impairment.
Extraarticular manifestations may develop during the clinical course of RA, even prior to the onset of arthritis (Fig. 380-2). Patients most likely to develop extraarticular disease have a history of smoking, have early onset of significant physical disability, and test positive for serum RF. Subcutaneous nodules, secondary Sjögren’s syndrome, pulmonary nodules, and anemia are among the most frequently observed extraarticular manifestations. Recent studies have shown a decrease in the incidence and severity of at least some extraarticular manifestations, particularly Felty’s syndrome and vasculitis.
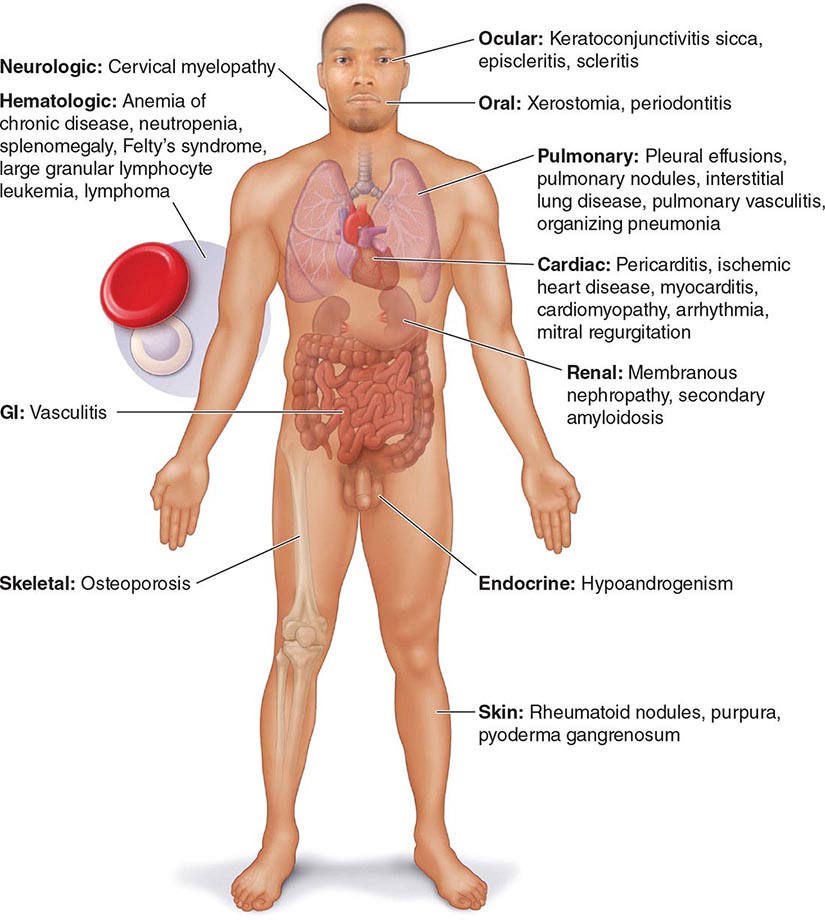
FIGURE 380-2 Extraarticular manifestations of rheumatoid arthritis.
The most common systemic and extraarticular features of RA are described in more detail in the sections below.
CONSTITUTIONAL
These signs and symptoms include weight loss, fever, fatigue, malaise, depression, and in the most severe cases, cachexia; they generally reflect a high degree of inflammation and may even precede the onset of joint symptoms. In general, the presence of a fever of >38.3°C (101°F) at any time during the clinical course should raise suspicion of systemic vasculitis (see below) or infection.
NODULES
Subcutaneous nodules occur in 30–40% of patients and more commonly in those with the highest levels of disease activity, the disease-related shared epitope (see below), a positive test for serum RF, and radiographic evidence of joint erosions. When palpated, the nodules are generally firm; nontender; and adherent to periosteum, tendons, or bursae; developing in areas of the skeleton subject to repeated trauma or irritation such as the forearm, sacral prominences, and Achilles tendon. They may also occur in the lungs, pleura, pericardium, and peritoneum. Nodules are typically benign, although they can be associated with infection, ulceration, and gangrene.
SJÖGREN’S SYNDROME
Secondary Sjögren’s syndrome (Chap. 383) is defined by the presence of either keratoconjunctivitis sicca (dry eyes) or xerostomia (dry mouth) in association with another connective tissue disease, such as RA. Approximately 10% of patients with RA have secondary Sjögren’s syndrome.
PULMONARY
Pleuritis, the most common pulmonary manifestation of RA, may produce pleuritic chest pain and dyspnea, as well as a pleural friction rub and effusion. Pleural effusions tend to be exudative with increased numbers of monocytes and neutrophils. Interstitial lung disease (ILD) may also occur in patients with RA and is heralded by symptoms of dry cough and progressive shortness of breath. ILD can be associated with cigarette smoking and is generally found in patients with higher disease activity, although it may be diagnosed in up to 3.5% of patients prior to the onset of joint symptoms. Diagnosis is readily made by high-resolution chest computed tomography (CT) scan. Pulmonary function testing shows a restrictive pattern (e.g., reduced total lung capacity) with a reduced diffusing capacity for carbon monoxide (DLCO). The presence of ILD confers a poor prognosis. The prognosis is not quite as poor as that of idiopathic pulmonary fibrosis (e.g., usual interstitial pneumonitis) because ILD secondary to RA responds more favorably than idiopathic ILD to immunosuppressive therapy (Chap. 315). Pulmonary nodules may be solitary or multiple. Caplan’s syndrome is a rare subset of pulmonary nodulosis characterized by the development of nodules and pneumoconiosis following silica exposure. Other less common pulmonary findings include respiratory bronchiolitis and bronchiectasis.
CARDIAC
The most frequent site of cardiac involvement in RA is the pericardium. However, clinical manifestations of pericarditis occur in less than 10% of patients with RA despite the fact that pericardial involvement may be detected in nearly one-half of the these patients by echocardiogram or autopsy studies. Cardiomyopathy, another clinically important manifestation of RA, may result from necrotizing or granulomatous myocarditis, coronary artery disease, or diastolic dysfunction. This involvement too may be subclinical and only identified by echocardiography or cardiac magnetic resonance imaging (MRI). Rarely, the heart muscle may contain rheumatoid nodules or be infiltrated with amyloid. Mitral regurgitation is the most common valvular abnormality in RA, occurring at a higher frequency than the general population.
VASCULITIS
Rheumatoid vasculitis (Chap. 385) typically occurs in patients with long-standing disease, a positive test for serum RF, and hypocomplementemia. The overall incidence has decreased significantly in the last decade to be less than 1% of patients. The cutaneous signs vary and include petechiae, purpura, digital infarcts, gangrene, livedo reticularis, and in severe cases large, painful lower extremity ulcerations. Vasculitic ulcers, which may be difficult to distinguish from those caused by venous insufficiency, may be treated successfully with immunosuppressive agents (requiring cytotoxic treatment in severe cases) as well as skin grafting. Sensorimotor polyneuropathies, such as mononeuritis multiplex, may occur in association with systemic rheumatoid vasculitis.
HEMATOLOGIC
A normochromic, normocytic anemia often develops in patients with RA and is the most common hematologic abnormality. The degree of anemia parallels the degree of inflammation, correlating with the levels of serum C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). Platelet counts may also be elevated in RA as an acute-phase reactant. Immune-mediated thrombocytopenia is rare in this disease.
Felty’s syndrome is defined by the clinical triad of neutropenia, splenomegaly, and nodular RA and is seen in less than 1% of patients, although its incidence appears to be declining in the face of more aggressive treatment of the joint disease. It typically occurs in the late stages of severe RA and is more common in whites than other racial groups. T cell large granular lymphocyte leukemia (T-LGL) may have a similar clinical presentation and often occurs in association with RA. T-LGL is characterized by a chronic, indolent clonal growth of LGL cells, leading to neutropenia and splenomegaly. As opposed to Felty’s syndrome, T-LGL may develop early in the course of RA. Leukopenia apart from these disorders is uncommon and most often due to drug therapy.
LYMPHOMA
Large cohort studies have shown a two- to fourfold increased risk of lymphoma in RA patients compared with the general population. The most common histopathologic type of lymphoma is a diffuse large B cell lymphoma. The risk of developing lymphoma increases if the patient has high levels of disease activity or Felty’s syndrome.
ASSOCIATED CONDITIONS
In addition to extraarticular manifestations, several conditions associated with RA contribute to disease morbidity and mortality rates. They are worthy of mention because they affect chronic disease management.
Cardiovascular Disease The most common cause of death in patients with RA is cardiovascular disease. The incidence of coronary artery disease and carotid atherosclerosis is higher in RA patients than in the general population even when controlling for traditional cardiac risk factors, such as hypertension, obesity, hypercholesterolemia, diabetes, and cigarette smoking. Furthermore, congestive heart failure (including both systolic and diastolic dysfunction) occurs at an approximately twofold higher rate in RA than in the general population. The presence of elevated serum inflammatory markers appears to confer an increased risk of cardiovascular disease in this population.
Osteoporosis Osteoporosis is more common in patients with RA than an age- and sex-matched population, with prevalence rates of 20–30%. The inflammatory milieu of the joint probably spills over into the rest of the body and promotes generalized bone loss by activating osteoclasts. Chronic use of glucocorticoids and disability-related immobility also contributes to osteoporosis. Hip fractures are more likely to occur in patients with RA and are significant predictors of increased disability and mortality rate in this disease.
Hypoandrogenism Men and postmenopausal women with RA have lower mean serum testosterone, luteinizing hormone (LH), and dehydroepiandrosterone (DHEA) levels than control populations. It has thus been hypothesized that hypoandrogenism may play a role in the pathogenesis of RA or arise as a consequence of the chronic inflammatory response. It is also important to realize that patients receiving chronic glucocorticoid therapy may develop hypoandrogenism owing to inhibition of LH and follicle-stimulating hormone (FSH) secretion from the pituitary gland. Because low testosterone levels may lead to osteoporosis, men with hypoandrogenism should be considered for androgen replacement therapy.
EPIDEMIOLOGY
RA affects approximately 0.5–1% of the adult population worldwide. There is evidence that the overall incidence of RA has been decreasing in recent decades, whereas the prevalence has remained the same because individuals with RA are living longer. The incidence and prevalence of RA varies based on geographic location, both globally and among certain ethnic groups within a country (Fig. 380-3). For example, the Native American Yakima, Pima, and Chippewa tribes of North America have reported prevalence rates in some studies of nearly 7%. In contrast, many population studies from Africa and Asia show lower prevalence rates for RA in the range of 0.2–0.4%.
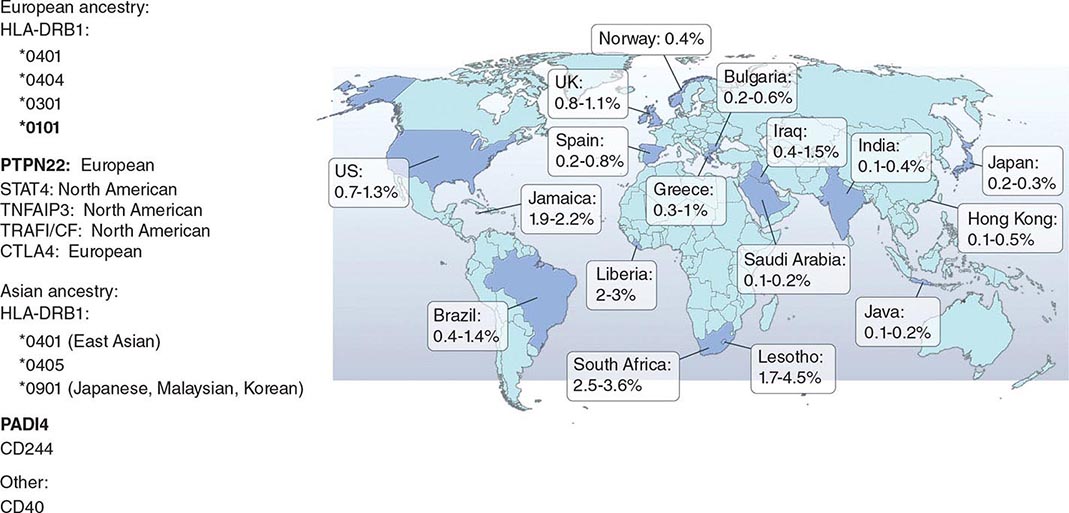
FIGURE 380-3 Global prevalence rates of rheumatoid arthritis (RA) with genetic associations. Listed are the major genetic alleles associated with RA. Although human leukocyte antigen (HLA)-DRB1 mutations are found globally, some alleles have been associated with RA in only certain ethnic groups.
Like many other autoimmune diseases, RA occurs more commonly in females than in males, with a 2–3:1 ratio. Interestingly, studies of RA from some of the Latin American and African countries show an even greater predominance of disease in females compared to males, with ratios of 6–8:1. Given this preponderance of females, various theories have been proposed to explain the possible role of estrogen in disease pathogenesis. Most of the theories center on the role of estrogens in enhancing the immune response. For example, some experimental studies have shown that estrogen can stimulate production of tumor necrosis factor a (TNF-α), a major cytokine in the pathogenesis of RA.
GENETIC CONSIDERATIONS
![]() It has been recognized for over 30 years that genetic factors contribute to the occurrence of RA as well as to its severity. The likelihood that a first-degree relative of a patient will share the diagnosis of RA is 2–10 times greater than in the general population. There remains, however, some uncertainty in the extent to which genetics plays a role in the causative mechanisms of RA. Although twin studies imply that genetic factors may explain up to 60% of the occurrence of RA, the more commonly stated estimate falls in the range of 10–25%. The estimate of genetic influence may vary across studies due to gene–environment interactions.
It has been recognized for over 30 years that genetic factors contribute to the occurrence of RA as well as to its severity. The likelihood that a first-degree relative of a patient will share the diagnosis of RA is 2–10 times greater than in the general population. There remains, however, some uncertainty in the extent to which genetics plays a role in the causative mechanisms of RA. Although twin studies imply that genetic factors may explain up to 60% of the occurrence of RA, the more commonly stated estimate falls in the range of 10–25%. The estimate of genetic influence may vary across studies due to gene–environment interactions.
The alleles known to confer the greatest risk of RA are located within the major histocompatibility complex (MHC). It has been estimated that one-third of the genetic risk for RA resides within this locus. Most, but probably not all, of this risk is associated with allelic variation in the HLA-DRB1 gene, which encodes the MHC II β-chain molecule. The disease-associated HLA-DRB1 alleles share an amino acid sequence at positions 70–74 in the third hypervariable regions of the HLA-DR β-chain, termed the shared epitope (SE). Carriership of the SE alleles is associated with production of anti-CCP antibodies and worse disease outcomes. Some of these HLA-DRB1 alleles bestow a high risk of disease (*0401), whereas others confer a more moderate risk (*0101, *0404, *1001, and *0901). Additionally, there is regional variation. In Greece, for example, where RA tends to be milder than in western European countries, RA susceptibility has been associated with the *0101 SE allele. By comparison, the *0401 or *0404 alleles are found in approximately 50–70% of northern Europeans and are the predominant risk alleles in this group. The most common disease susceptibility SE alleles in Asians, namely the Japanese, Koreans, and Chinese, are *0405 and *0901. Lastly, disease susceptibility of Native American populations such as the Pima and Tlingit Indians, where the prevalence of RA can be as high as 7%, is associated with the SE allele *1042. The risk of RA conferred by these SE alleles is less in African and Hispanic Americans than in individuals of European ancestry.
Genome-wide association studies (GWAS) have made possible the identification of several non-MHC-related genes that contribute to RA susceptibility. GWAS are based on the detection of single-nucleotide polymorphisms (SNPs), which allow for examination of the genetic architecture of complex diseases such as RA. There are approximately 10 million common SNPs within a human genome consisting of 3 billion base pairs. As a rule, GWAS identify only common variants, namely, those with a frequency of more than 5% in the general population.
Overall, several themes have emerged from GWAS in RA. First, the non-MHC loci identified as risk alleles for RA have only a modest effect on risk; they also contribute to the risk for developing other autoimmune diseases, such as type 1 diabetes mellitus, systemic lupus erythematosus, and multiple sclerosis. Second, although most of the non-HLA associations are described in patients with anti-CCP antibody-positive disease, there are several risk loci that are unique to anti-CCP antibody-negative disease. Third, risk alleles vary among ethnic groups. And fourth, the risk loci mostly reside in genes encoding proteins involved in the regulation of the immune response. However, the risk alleles identified by GWAS only account at present for approximately 5% of the genetic risk, suggesting that rare variants or other classes of DNA variants, such as variants in copy number, may be yet found that significantly contribute to the overall risk model.
Recently, imputation of SNP data from a GWAS meta-analysis shows amino acid substitutions in the MHC locus independently associated with the risk for RA are at position 11, 71, and 74 in HLA-DRβ1, position 9 of HLA-B, and position 9 of HLA-DPβ1. The amino acids at position 11, 71, and 74 are located in the antigen-binding grove of the HLA-DRβ1 molecule, highlighting positions 71 and 74 that form part of the original shared epitope.
Among the best examples of the non-MHC genes contributing to the risk of RA is the gene encoding protein tyrosine phosphatase non-receptor 22 (PTPN22). This gene varies in frequency among patients from different parts of Europe (e.g., 3–10%), but is absent in patients of East Asian ancestry. PTPN22 encodes lymphoid tyrosine phosphatase, a protein that regulates T and B cell function. Inheritance of the risk allele for PTPN22 produces a gain-of-function in the protein that is hypothesized to result in the abnormal thymic selection of autoreactive T and B cells and appears to be associated exclusively with anti-CCP-positive disease. The peptidyl arginine deiminase type IV (PADI4) gene is another risk allele that encodes an enzyme involved in the conversion of arginine to citrulline and is postulated to play a role in the development of antibodies to citrullinated antigens. A polymorphism in PADI4 has been associated with RA only in Asian populations.
Epigenetics is the study of heritable traits that affect gene expression but do not modify DNA sequence. It may provide a link between environmental exposure and predisposition to disease. The best-studied mechanisms include posttranslational histone modifications and DNA methylation. Although studies of epigenetic phenomena are limited, DNA methylation patterns have been shown to differ between RA patients and healthy controls, as well as patients with osteoarthritis.
ENVIRONMENTAL FACTORS
In addition to genetic predisposition, a host of environmental factors have been implicated in the pathogenesis of RA. The most reproducible of these environmental links is cigarette smoking. Numerous cohort and case control studies have demonstrated that smoking confers a relative risk for developing RA of 1.5–3.5. In particular, women who smoke cigarettes have a nearly 2.5 times greater risk of RA, a risk that persists even 15 years after smoking cessation. A twin who smokes will have a significantly higher risk for RA than his or her monozygotic co-twin, theoretically with the same genetic risk, who does not smoke. Interestingly, the risk from smoking is almost exclusively related to RF and anti-CCP antibody-positive disease. However, it has not been shown that smoking cessation, while having many health benefits, improves disease activity.
Researchers began to aggressively seek an infectious etiology for RA after the discovery in 1931 that sera from patients with this disease could agglutinate strains of streptococci. Certain viruses such as Epstein-Barr virus (EBV) have garnered the most interest over the past 30 years given their ubiquity, ability to persist for many years in the host, and frequent association with arthritic complaints. For example, titers of IgG antibodies against EBV antigens in the peripheral blood and saliva are significantly higher in patients with RA than the general population. EBV DNA has also been found in synovial fluid and synovial cells of RA patients. Because the evidence for these links is largely circumstantial, it has not been possible to directly implicate infection as a causative factor in RA.
PATHOLOGY
RA affects the synovial tissue and underlying cartilage and bone. The synovial membrane, which covers most articular surfaces, tendon sheaths, and bursae, normally is a thin layer of connective tissue. In joints, it faces the bone and cartilage, bridging the opposing bony surfaces and inserting at periosteal regions close to the articular cartilage. It consists primarily of two cell types—type A synoviocytes (macrophage-derived) and type B synoviocytes (fibroblast-derived). The synovial fibroblasts are the most abundant and produce the structural components of joints, including collagen, fibronectin, and laminin, as well as other extracellular constituents of the synovial matrix. The sublining layer consists of blood vessels and a sparse population of mononuclear cells within a loose network of connective tissue. Synovial fluid, an ultrafiltrate of blood, diffuses through the subsynovial lining tissue across the synovial membrane and into the joint cavity. Its main constituents are hyaluronan and lubricin. Hyaluronan is a glycosaminoglycan that contributes to the viscous nature of synovial fluid, which along with lubricin, lubricates the surface of the articular cartilage.
The pathologic hallmarks of RA are synovial inflammation and proliferation, focal bone erosions, and thinning of articular cartilage. Chronic inflammation leads to synovial lining hyperplasia and the formation of pannus, a thickened cellular membrane containing fibroblast-like synoviocytes and granulation-reactive fibrovascular tissue that invades the underlying cartilage and bone. The inflammatory infiltrate is made up of no less than six cell types: T cells, B cells, plasma cells, dendritic cells, mast cells, and, to a lesser extent, granulocytes. The T cells comprise 30–50% of the infiltrate, with the other cells accounting for the remainder. The topographical organization of these cells is complex and may vary among individuals with RA. Most often, the lymphocytes are diffusely organized among the tissue resident cells; however, in some cases, the B cells, T cells, and dendritic cells may form higher levels of organization, such as lymphoid follicles and germinal center–like structures. Growth factors secreted by synovial fibroblasts and macrophages promote the formation of new blood vessels in the synovial sublining that supply the increasing demands for oxygenation and nutrition required by the infiltrating leukocytes and expanding synovial tissue.
The structural damage to the mineralized cartilage and subchondral bone is mediated by the osteoclast. Osteoclasts are multinucleated giant cells that can be identified by their expression of CD68, tartrate-resistant acid phosphatase, cathepsin K, and the calcitonin receptor. They appear at the pannus-bone interface where they eventually form resorption lacunae. These lesions typically localize where the synovial membrane inserts into the periosteal surface at the edges of bones close to the rim of articular cartilage and at the attachment sites of ligaments and tendon sheaths. This process most likely explains why bone erosions usually develop at the radial sites of the MCP joints juxtaposed to the insertion sites of the tendons, collateral ligaments, and synovial membrane. Another form of bone loss is periarticular osteopenia that occurs in joints with active inflammation. It is associated with substantial thinning of the bony trabeculae along the metaphyses of bones, and likely results from inflammation of the bone marrow cavity. These lesions can be visualized on MRI scans, where they appear as signal alterations in the bone marrow adjacent to inflamed joints. Their signal characteristics show they are water-rich with a low fat content and are consistent with highly vascularized inflammatory tissue. These bone marrow lesions are often the forerunner of bone erosions.
The cortical bone layer that separates the bone marrow from the invading pannus is relatively thin and susceptible to penetration by the inflamed synovium. The bone marrow lesions seen on MRI scans are associated with an endosteal bone response characterized by the accumulation of osteoblasts and deposition of osteoid. Thus, in recent years, the concept of joint pathology in RA has been extended to include the bone marrow cavity. Finally, generalized osteoporosis, which results in the thinning of trabecular bone throughout the body, is a third form of bone loss found in patients with RA.
Articular cartilage is an avascular tissue comprised of a specialized matrix of collagens, proteoglycans, and other proteins. It is organized in four distinct regions (superficial, middle, deep, and calcified cartilage zones)—chondrocytes constitute the unique cellular component in these layers. Originally, cartilage was considered to be an inert tissue, but it is now known to be a highly responsive tissue that reacts to inflammatory mediators and mechanical factors, which in turn, alter the balance between cartilage anabolism and catabolism. In RA, the initial areas of cartilage degradation are juxtaposed to the synovial pannus. The cartilage matrix is characterized by a generalized loss of proteoglycan, most evident in the superficial zones adjacent to the synovial fluid. Degradation of cartilage may also take place in the perichondrocytic zone and in regions adjacent to the subchondral bone.
PATHOGENESIS
The pathogenic mechanisms of synovial inflammation are likely to result from a complex interplay of genetic, environmental, and immunologic factors that produces dysregulation of the immune system and a breakdown in self-tolerance (Fig. 380-4). Precisely what triggers these initiating events and what genetic and environmental factors disrupt the immune system remains a mystery. However, a detailed molecular picture is emerging of the mechanisms underlying the chronic inflammatory response and the destruction of the articular cartilage and bone.
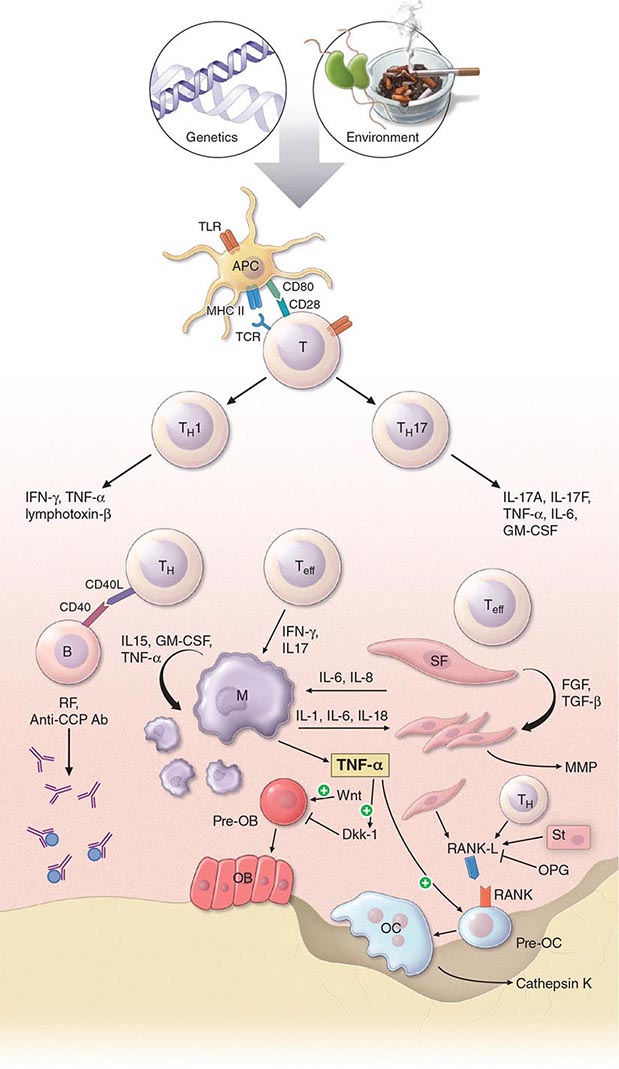
FIGURE 380-4 Pathophysiologic mechanisms of inflammation and joint destruction. Genetic predisposition along with environmental factors may trigger the development of rheumatoid arthritis (RA), with subsequent synovial T cell activation. CD4+ T cells become activated by antigen-presenting cells (APCs) through interactions between the T cell receptor and class II major histocompatibility complex (MHC)-peptide antigen (signal 1) with co-stimulation through the CD28-CD80/86 pathway, as well as other pathways (signal 2). In theory, ligands binding Toll-like receptors (TLRs) may further stimulate activation of APCs inside the joint. Synovial CD4+ T cells differentiate into TH1 and TH17 cells, each with their distinctive cytokine profile. CD4+ TH cells in turn activate B cells, some of which are destined to differentiate into autoantibody-producing plasma cells. Immune complexes, possibly comprised of rheumatoid factors (RFs) and anti–cyclic citrullinated peptides (CCP) antibodies, may form inside the joint, activating the complement pathway and amplifying inflammation. T effector cells stimulate synovial macrophages (M) and fibroblasts (SF) to secrete proinflammatory mediators, among which is tumor necrosis factor α (TNF-α). TNF-α upregulates adhesion molecules on endothelial cells, promoting leukocyte influx into the joint. It also stimulates the production of other inflammatory mediators, such as interleukin 1 (IL-1), IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF). TNF-α has a critically important function in regulating the balance between bone destruction and formation. It upregulates the expression of dickkopf-1 (DKK-1), which can then internalize Wnt receptors on osteoblast precursors. Wnt is a soluble mediator that promotes osteoblastogenesis and bone formation. In RA, bone formation is inhibited through the Wnt pathway, presumably due to the action of elevated levels of DKK-1. In addition to inhibiting bone formation, TNF-α stimulates osteoclastogenesis. However, it is not sufficient by itself to induce the differentiation of osteoclast precursors (Pre-OC) into activated osteoclasts capable of eroding bone. Osteoclast differentiation requires the presence of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB (RANK) ligand (RANKL), which binds to RANK on the surface of Pre-OC. Inside the joint, RANKL is mainly derived from stromal cells, synovial fibroblasts, and T cells. Osteoprotegerin (OPG) acts as a decoy receptor for RANKL, thereby inhibiting osteoclastogenesis and bone loss. FGF, fibroblast growth factor; IFN, interferon; TGF, transforming growth factor.
In RA, the preclinical stage appears to be characterized by a breakdown in self-tolerance. This idea is supported by the finding that autoantibodies, such as RF and anti-CCP antibodies, may be found in sera from patients many years before clinical disease can be detected. However, the antigenic targets of anti-CCP antibodies and RF are not restricted to the joint, and their role in disease pathogenesis remains speculative. Anti-CCP antibodies are directed against deaminated peptides, which result from posttranslational modification by the enzyme PADI4. They recognize citrulline-containing regions of several different matrix proteins, including filaggrin, keratin, fibrinogen, and vimentin, and are present at higher levels in the joint fluid compared to the serum. Other autoantibodies have been found in a minority of patients with RA, but they also occur in the setting of other types of arthritis. They bind to a diverse array of autoantigens, including type II collagen, human cartilage gp-39, aggrecan, calpastatin, BiP (immunoglobulin binding protein), and glucose-6-phosphate isomerase.
In theory, environmental stimulants may synergize with other factors to bring about inflammation in RA. People who smoke display higher citrullination of proteins in bronchoalveolar fluid than those who do not smoke. Thus, it has been speculated that long-term exposure to tobacco smoke might induce citrullination of cellular proteins in the lung and stimulate the expression of a neoepitope capable of inducing self-reactivity, which in turns, leads to formation of immune complexes and joint inflammation. Exposure to silicone dust and mineral oil, which has adjuvant effects, has also been linked to an increased risk for anti-CCP antibody-positive RA.
How might microbes or their products be involved in the initiating events of RA? The immune system is alerted to the presence of microbial infections through Toll-like receptors (TLRs). There are 10 TLRs in humans that recognize a variety of microbial products, including bacterial cell-surface lipopolysaccharides and heat-shock proteins (TLR4), lipoproteins (TLR2), double-strand RNA viruses (TLR3), and unmethylated CpG DNA from bacteria (TLR9). TLR2, -3, and -4 are abundantly expressed by synovial fibroblasts in early RA and, when bound by their ligands, upregulate production of proinflammatory cytokines. Although such events could amplify inflammatory pathways in RA, a specific role for TLRs in disease pathogenesis has not been elucidated.
The pathogenesis of RA is built upon the concept that self-reactive T cells drive the chronic inflammatory response. In theory, self-reactive T cells might arise in RA from abnormal central (thymic) selection due to defects in DNA repair leading to an imbalance of T cell death and life, or defects in the cell signaling apparatus lowering the threshold for T cell activation. Similarly, abnormal selection of the T cell repertoire in the periphery might lead to a breakdown in T cell tolerance. The support for these theories comes mainly from studies of arthritis in mouse models. It has not been shown that patients with RA have abnormal thymic selection of T cells or defective apoptotic pathways regulating cell death. At least some antigen stimulation inside the joint seems likely, owing to the fact that T cells in the synovium express a cell-surface phenotype indicating prior antigen exposure and show evidence of clonal expansion. Of interest, peripheral blood T cells from patients with RA have been shown to display a fingerprint of premature aging that mostly affects inexperienced naïve T cells. In these studies, the most glaring findings have been the loss of telomeric sequences and a decrease in the thymic output of new T cells. Although intriguing, it is not clear how generalized T cell abnormalities might provoke a systemic disease dominated by synovitis.
There is substantial evidence supporting a role for CD4+ T cells in the pathogenesis of RA. First, the co-receptor CD4 on the surface of T cells binds to invariant sites on MHC class II molecules, stabilizing the MHC-peptide–T cell receptor complex during T cell activation. Because the SE on MHC class II molecules is a risk factor for RA, it follows that CD4+ T cell activation may play a role in the pathogenesis of this disease. Second, CD4+ memory T cells are enriched in the synovial tissue from patients with RA and can be implicated through “guilt by association.” Third, CD4+ T cells have been shown to be important in the initiation of arthritis in animal models. Fourth, some, but not all, T cell–directed therapies have shown clinical efficacy in this disease. Taken together, these lines of evidence suggest that CD4+ T cells play an important role in orchestrating the chronic inflammatory response in RA. However, other cell types, such as CD8+ T cells, natural killer (NK) cells, and B cells are present in synovial tissue and may also influence pathogenic responses.
In the rheumatoid joint, by mechanisms of cell-cell contact and release of soluble mediators, activated T cells stimulate macrophages and fibroblast-like synoviocytes to generate proinflammatory mediators and proteases that drive the synovial inflammatory response and destroy the cartilage and bone. CD4+ T cell activation is dependent on two signals: (1) T cell receptor binding to peptide-MHC on antigen-presenting cells; and (2) CD28 binding to CD80/86 on antigen-presenting cells. CD4+ T cells also provide help to B cells, which in turn, produce antibodies that may promote further inflammation in the joint. The previous T cell–centric model for the pathogenesis of RA was based on a TH1-driven paradigm, which came from studies indicating that CD4+ T helper (TH) cells differentiated into TH1 and TH2 subsets, each with their distinctive cytokine profiles. TH1 cells were found to mainly produce interferon γ (IFN-γ), lymphotoxin β, and TNF-α, whereas TH2 cells predominately secreted interleukin (IL)-4, IL-5, IL-6, IL-10, and IL-13. The recent discovery of another subset of TH cells, namely the TH17 lineage, has revolutionized our concepts concerning the pathogenesis of RA. In humans, naïve T cells are induced to differentiate into TH17 cells by exposure to transforming growth factor β (TGF-β), IL-1, IL-6, and IL-23. Upon activation, TH17 cells secrete a variety of proinflammatory mediators such as IL-17, IL-21, IL-22, TNF-α, IL-26, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF). Substantial evidence now exists from both animal models and humans that IL-17 plays an important role not only in promoting joint inflammation, but also in destroying cartilage and subchondral bone
The immune system has evolved mechanisms to counterbalance the potential harmful immune-mediated inflammatory responses provoked by infectious agents and other triggers. Among these negative regulators are regulatory T (Treg) cells, which are produced in the thymus and induced in the periphery to suppress immune-mediated inflammation. They are characterized by the surface expression of CD25 and the transcription factor forkhead box P3 (FOXP3) and orchestrate dominant tolerance through contact with other immune cells and secretion of inhibitory cytokines, such as TGF-β, IL-10, and IL-35. Treg cells appear to be heterogeneous and capable of suppressing distinct classes (TH1, TH2, TH17) of the immune response. In RA, the data that Treg numbers are deficient compared to normal healthy controls are contradictory and inconclusive. Although some experimental evidence suggests that Treg suppressive activity is lost due to dysfunctional expression of cytotoxic T lymphocyte antigen 4 (CTLA-4), the nature of Treg defects in RA, if they exist, remains unclear.
Cytokines, chemokines, antibodies, and endogenous danger signals bind to receptors on the surface of immune cells and stimulate a cascade of intracellular signaling events that can amplify the inflammatory response. Signaling molecules and their binding partners in these pathways are the target of small-molecule drugs designed to interfere with signal transduction and block these reinforcing inflammatory loops. Examples of signal molecules in these critical inflammatory pathways include Janus kinase (JAK)/signal transducers and activators of transcription (STAT), spleen tyrosine kinase (Syk), mitogen-activated protein kinases (MAPKs), and nuclear factor-κB (NF-κB). These pathways exhibit significant cross-talk and are found in many cell types. Some signal transducers, such as JAK3, are primarily expressed in hematopoietic cells and play an important role in the inflammatory response in RA.
Activated B cells are also important players in the chronic inflammatory response. B cells give rise to plasma cells, which in turn, produce antibodies, including RF and anti-CCP antibodies. RFs may form large immune complexes inside the joint that contribute to the pathogenic process by fixing complement and promoting the release of proinflammatory chemokines and chemoattractants. In mouse models of arthritis, RF-containing immune complexes and anti-CCP-containing immune complexes synergize with other mechanisms to exacerbate the synovial inflammatory response.
RA is often considered to be a macrophage-driven disease because this cell type is the predominant source of proinflammatory cytokines inside the joint. Key proinflammatory cytokines released by synovial macrophages include TNF-α, IL-1, IL-6, IL-12, IL-15, IL-18, and IL-23. Synovial fibroblasts, the other major cell type in this microenvironment, produce the cytokines IL-1 and IL-6 as well as TNF-α. TNF-α is a pivotal cytokine in the pathobiology of synovial inflammation. It upregulates adhesion molecules on endothelial cells, promoting the influx of leukocytes into the synovial microenvironment; activates synovial fibroblasts; stimulates angiogenesis; promotes pain receptor sensitizing pathways; and drives osteoclastogenesis. Fibroblasts secrete matrix metalloproteinases (MMPs) as well as other proteases that are chiefly responsible for the breakdown of articular cartilage.
Osteoclast activation at the site of the pannus is closely tied to the presence of focal bone erosion. Receptor activator of nuclear factor-κB ligand (RANKL) is expressed by stromal cells, synovial fibroblasts, and T cells. Upon binding to its receptor RANK on osteoclast progenitors, RANKL stimulates osteoclast differentiation and bone resorption. RANKL activity is regulated by osteoprotegerin (OPG), a decoy receptor of RANKL that blocks osteoclast formation. Monocytic cells in the synovium serve as the precursors of osteoclasts and, when exposed to macrophage colony-stimulating factor (M-CSF) and RANKL, fuse to form polykaryons termed preosteoclasts. These precursor cells undergo further differentiation into osteoclasts with the characteristic ruffled membrane. Cytokines such as TNF-α, IL-1, IL-6, and IL-17 increase the expression of RANKL in the joint and thus promote osteoclastogenesis. Osteoclasts also secrete cathepsin K, which is a cysteine protease that degrades the bone matrix by cleaving collagen. Stimulation of osteoclasts also contributes to generalized bone loss and osteoporosis.
Increased bone loss is only part of the story in RA, as decreased bone formation plays a crucial role in bone remodeling at sites of inflammation. Recent evidence shows that inflammation suppresses bone formation. The proinflammatory cytokine TNF-α plays a key role in actively suppressing bone formation by enhancing the expression of dickkopf-1 (DKK-1). DKK-1 is an important inhibitor of the Wnt pathway, which acts to promote osteoblast differentiation and bone formation. The Wnt system is a family of soluble glycoproteins that bind to cell-surface receptors known as frizzled (fz) and low-density lipoprotein (LDL) receptor–related proteins (LRPs) and promote cell growth. In animal models, increased levels of DKK-1 are associated with decreased bone formation, whereas inhibition of DKK-1 protects against structural damage in the joint. Wnt proteins also induce the formation of OPG and thereby shut down bone resorption, emphasizing their key role in tightly regulating the balance between bone resorption and formation.
DIAGNOSIS
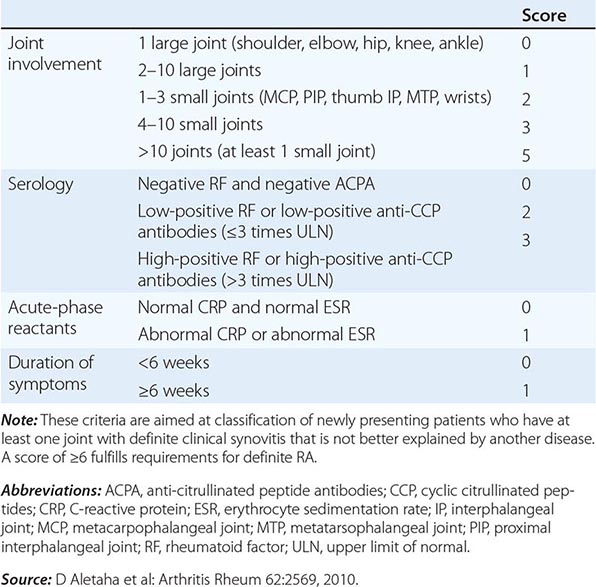
The clinical diagnosis of RA is largely based on signs and symptoms of a chronic inflammatory arthritis, with laboratory and radiographic results providing important supplemental information. In 2010, a collaborative effort between the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) revised the 1987 ACR classification criteria for RA in an effort to improve early diagnosis with the goal of identifying patients who would benefit from early introduction of disease-modifying therapy (Table 380-1). Application of the newly revised criteria yields a score of 0–10, with a score of ≥ 6 fulfilling the requirements for definite RA. The new classification criteria differ in several ways from the older criteria set. The new criteria include a positive test for serum anti-CCP antibodies (also termed ACPA, anti-citrullinated peptide antibodies) as an item, which carries greater specificity for the diagnosis of RA than a positive test for RF. The newer classification criteria also do not take into account whether the patient has rheumatoid nodules or radiographic joint damage because these findings occur rarely in early RA. It is important to emphasize that the new 2010 ACR-EULAR criteria are “classification criteria” as opposed to “diagnostic criteria” and serve to distinguish patients at the onset of disease who have a high likelihood of evolution to chronic disease with persistent synovitis and joint damage. The presence of radiographic joint erosions or subcutaneous nodules may inform the diagnosis in the later stages of the disease.
CLASSIFICATION CRITERIA FOR RHEUMATOID ARTHRITIS |
LABORATORY FEATURES
Patients with systemic inflammatory diseases such as RA will often present with elevated nonspecific inflammatory markers such as an ESR or CRP. Detection of serum RF and anti-CCP antibodies is important in differentiating RA from other polyarticular diseases, although RF lacks diagnostic specificity and may be found in association with other chronic inflammatory diseases in which arthritis figures in the clinical manifestations.
IgM, IgG, and IgA isotypes of RF occur in sera from patients with RA, although the IgM isotype is the one most frequently measured by commercial laboratories. Serum IgM RF has been found in 75–80% of patients with RA; therefore, a negative result does not exclude the presence of this disease. It is also found in other connective tissue diseases, such as primary Sjögren’s syndrome, systemic lupus erythematosus, and type II mixed essential cryoglobulinemia, as well as chronic infections such as subacute bacterial endocarditis and hepatitis B and C. Serum RF may also be detected in 1–5% of the healthy population.
The presence of serum anti-CCP antibodies has about the same sensitivity as serum RF for the diagnosis of RA. However, its diagnostic specificity approaches 95%, so a positive test for anti-CCP antibodies in the setting of an early inflammatory arthritis is useful for distinguishing RA from other forms of arthritis. There is some incremental value in testing for the presence of both RF and anti-CCP, as some patients with RA are positive for RF but negative for anti-CCP and visa versa. The presence of RF or anti-CCP antibodies also has prognostic significance, with anti-CCP antibodies showing the most value for predicting worse outcomes.
SYNOVIAL FLUID ANALYSIS
Typically, synovial fluid from patients with RA reflects an inflammatory state. Synovial fluid white blood cell (WBC) counts can vary widely, but generally range between 5000 and 50,000 WBC/μL compared to <2000 WBC/μL for a noninflammatory condition such as osteoarthritis. In contrast to the synovial tissue, the overwhelming cell type in the synovial fluid is the neutrophil. Clinically, the analysis of synovial fluid is most useful for confirming an inflammatory arthritis (as opposed to osteoarthritis), while at the same time excluding infection or a crystal-induced arthritis such as gout or pseudogout (Chap. 395).
JOINT IMAGING
Joint imaging is a valuable tool not only for diagnosing RA, but also for tracking progression of any joint damage. Plain x-ray is the most common imaging modality, but it is limited to visualization of the bony structures and inferences about the state of the articular cartilage based on the amount of narrowing of the joint space. MRI and ultrasound techniques offer the added value of detecting changes in the soft tissues such as synovitis, tenosynovitis, and effusions as well as greater sensitivity for identifying bony abnormalities. Plain radiographs are usually relied upon in clinical practice for the purpose of diagnosis and monitoring of affected joints. However, in selected cases, MRI and ultrasound can provide additional diagnostic information that may guide clinical decision making. Musculoskeletal ultrasound with power Doppler is increasingly used in rheumatology clinical practice for detecting synovitis and bone erosion.
Plain Radiography Classically in RA, the initial radiographic finding is periarticular osteopenia. Practically speaking, however, this finding is difficult to appreciate on plain films and, in particular, on the newer digitalized x-rays. Other findings on plain radiographs include soft tissue swelling, symmetric joint space loss, and subchondral erosions, most frequently in the wrists and hands (MCPs and PIPs) and the feet (MTPs). In the feet, the lateral aspect of the fifth MTP is often targeted first, but other MTP joints may be involved at the same time. X-ray imaging of advanced RA may reveal signs of severe destruction, including joint subluxation and collapse (Fig. 380-5).
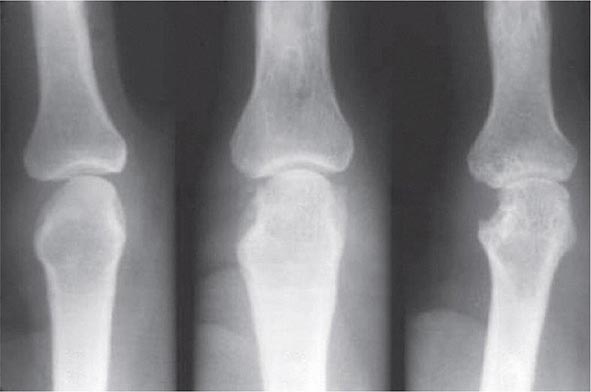
FIGURE 380-5 X-ray demonstrating progression of erosions on the proximal interphalangeal joint. (Courtesy of the American College of Rheumatology.)
MRI MRI offers the greatest sensitivity for detecting synovitis and joint effusions, as well as early bone and bone marrow changes. These soft tissue abnormalities often occur before osseous changes are noted on x-ray. Presence of bone marrow edema has been recognized to be an early sign of inflammatory joint disease and can predict the subsequent development of erosions on plain radiographs as well as MRI scans. Cost and availability of MRI are the main factors limiting its routine clinical use.
Ultrasound Ultrasound, including power color Doppler, has the ability to detect more erosions than plain radiography, especially in easily accessible joints. It can also reliably detect synovitis, including increased joint vascularity indicative of inflammation. The usefulness of ultrasound is dependent on the experience of the sonographer; however, it does offer the advantages of portability, lack of radiation, and low expense relative to MRI, factors that make it attractive as a clinical tool.
CLINICAL COURSE
The natural history of RA is complex and affected by a number of factors including age of onset, gender, genotype, phenotype (i.e., extraarticular manifestations or variants of RA), and comorbid conditions, which make for a truly heterogeneous disease. There is no simple way to predict the clinical course. It is important to realize that as many as 10% of patients with inflammatory arthritis fulfilling ACR classification criteria for RA will undergo a spontaneous remission within 6 months (particularly seronegative patients). However, the vast majority of patients will exhibit a pattern of persistent and progressive disease activity that waxes and wanes in intensity over time. A minority of patients will show intermittent and recurrent explosive attacks of inflammatory arthritis interspersed with periods of disease quiescence. Finally, an aggressive form of RA may occur in an unfortunate few with inexorable progression of severe erosive joint disease, although this highly destructive course is less common in the modern treatment era of biologics.
Disability, as measured by the Health Assessment Questionnaire (HAQ), shows gradual worsening of disability over time in the face of poorly controlled disease activity and disease progression. Disability may result from both a disease activity–related component that is potentially reversible with therapy and a joint damage–related component owing to the cumulative and largely irreversible effects of cartilage and bone breakdown. Early in the course of disease, the extent of joint inflammation is the primary determinant of disability, while in the later stages of disease, the amount of joint damage is the dominant contributing factor. Previous studies have shown that more than one-half of patients with RA are unable to work 10 years after the onset of their disease; however, increased employability and less work absenteeism has been reported recently with the use of newer therapies and earlier treatment intervention.
The overall mortality rate in RA is two times greater than the general population, with ischemic heart disease being the most common cause of death followed by infection. Median life expectancy is shortened by an average of 7 years for men and 3 years for women compared to control populations. Patients at higher risk for shortened survival are those with systemic extraarticular involvement, low functional capacity, low socioeconomic status, low education, and chronic prednisone use.
TREATMENT | RHEUMATOID ARTHRITIS |
The amount of clinical disease activity in patients with RA reflects the overall burden of inflammation and is the variable most influencing treatment decisions. Joint inflammation is the main driver of joint damage and is the most important cause of functional disability in the early stages of disease. Several composite indices have been developed to assess clinical disease activity. The ACR 20, 50, and 70 improvement criteria (which corresponds to a 20%, 50%, and 70% improvement, respectively, in joint counts, physician/patient assessment of disease severity, pain scale, serum levels of acute-phase reactants [ESR or CRP], and a functional assessment of disability using a self-administered patient questionnaire) are a composite index with a dichotomous response variable. The ACR improvement criteria are commonly used in clinical trials as an endpoint for comparing the proportion of responders between treatment groups. In contrast, the Disease Activity Score (DAS), Simplified Disease Activity Index (SDAI), and the Clinical Disease Activity Index (CDAI) are continuous measures of disease activity. These scales are increasingly used in clinical practice for tracking disease status and, in particular, for documenting treatment response.
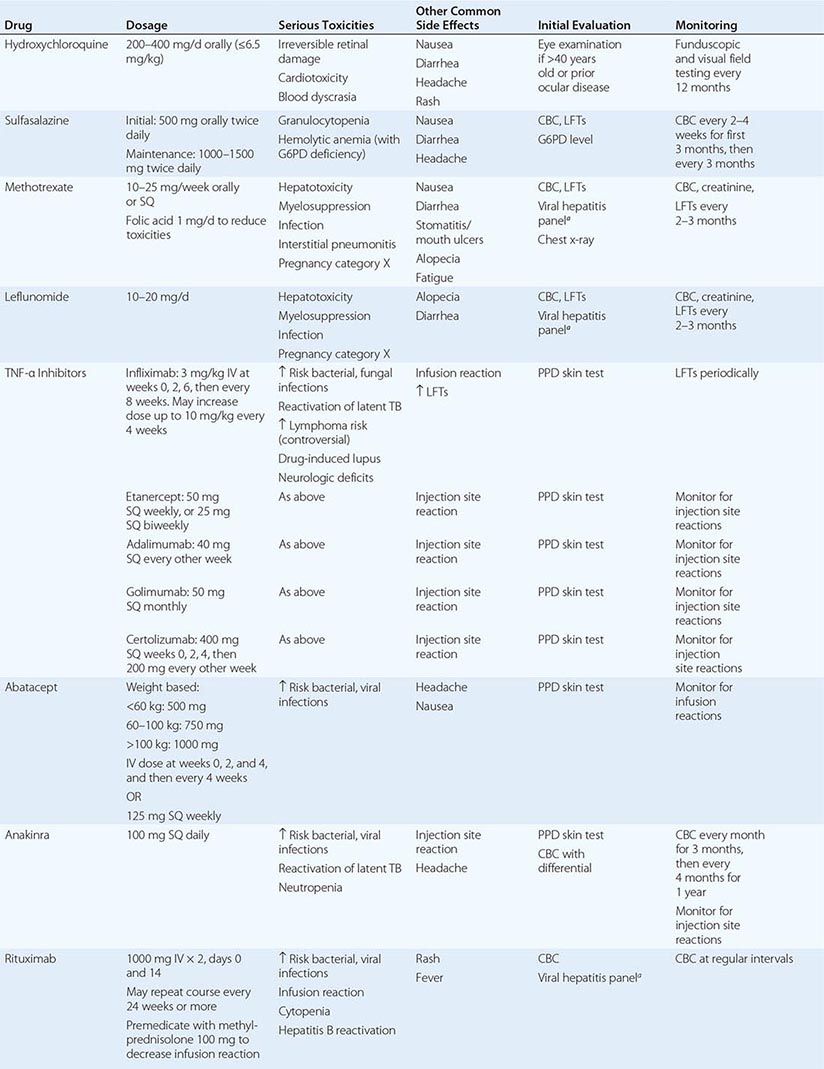
Several developments during the past two decades have changed the therapeutic landscape in RA. They include (1) the emergence of methotrexate as the disease-modifying antirheumatic drug (DMARD) of first choice for the treatment of early RA; (2) the development of novel highly efficacious biologicals that can be used alone or in combination with methotrexate; and (3) the proven superiority of combination DMARD regimens over methotrexate alone. The medications used for the treatment of RA may be divided into broad categories: nonsteroidal anti-inflammatory drugs (NSAIDs); glucocorticoids, such as prednisone and methylprednisolone; conventional DMARDs; and biologic DMARDs (Table 380-2). Although disease for some patients with RA is managed adequately with a single DMARD, such as methotrexate, the situation in most cases demands the use of a combination DMARD regimen that may vary in its components over the treatment course depending on fluctuations in disease activity and emergence of drug-related toxicities and comorbidities.
DMARDs USED FOR THE TREATMENT OF RHEUMATOID ARTHRITIS |
NSAIDs
NSAIDs were formerly viewed as the core of all other RA therapy, but they are now considered to be adjunctive therapy for management of symptoms uncontrolled by other measures. NSAIDs exhibit both analgesic and anti-inflammatory properties. The anti-inflammatory effects of NSAIDs derive from their ability to nonselectively inhibit cyclooxygenase (COX)-1 and COX-2. Although the results of clinical trials suggest NSAIDs are roughly equivalent in their efficacy, experience suggests that some individuals may preferentially respond to a particular NSAID. Chronic use should be minimized due to the possibility of side effects, including gastritis and peptic ulcer disease as well as impairment of renal function.
GLUCOCORTICOIDS
Glucocorticoids may serve in several ways to control disease activity in RA. First, they may be administered in low to moderate doses to achieve rapid disease control before the onset of fully effective DMARD therapy, which often takes several weeks or even months. Second, a 1- to 2-week burst of glucocorticoids may be prescribed for the management of acute disease flares, with dose and duration guided by the severity of the exacerbation. Chronic administration of low doses (5–10 mg/d) of prednisone (or its equivalent) may also be warranted to control disease activity in patients with an inadequate response to DMARD therapy. Low-dose prednisone therapy has been shown in prospective studies to retard radiographic progression of joint disease; however, the benefits of this approach must be carefully weighed against the risks. Best practices minimize chronic use of low-dose prednisone therapy owing to the risk of osteoporosis and other long-term complications; however, the use of chronic prednisone therapy is unavoidable in many cases. High-dose glucocorticoids may be necessary for treatment of severe extraarticular manifestations of RA, such as ILD. Finally, if a patient exhibits one or a few actively inflamed joints, the clinician may consider intraarticular injection of an intermediate-acting glucocorticoid such as triamcinolone acetonide. This approach may allow for rapid control of inflammation in the setting of a limited number of affected joints. Caution must be exercised to appropriately exclude joint infection, as it often mimics an RA flare.
Osteoporosis ranks as an important long-term complication of chronic prednisone use. The ACR recommends primary prevention of glucocorticoid-induced osteoporosis with a bisphosphonate in any patient receiving 5 mg/d or more of prednisone for greater than 3 months. Although prednisone use is known to increase the risk of peptic ulcer disease, especially with concomitant NSAID use, no evidence-based guidelines have been published regarding the use of gastrointestinal ulcer prophylaxis in this situation.
DMARDs
DMARDs are so named because of their ability to slow or prevent structural progression of RA. The conventional DMARDs include hydroxychloroquine, sulfasalazine, methotrexate, and leflunomide; they exhibit a delayed onset of action of approximately 6–12 weeks. Methotrexate is the DMARD of choice for the treatment of RA and is the anchor drug for most combination therapies. It was approved for the treatment of RA in 1986 and remains the benchmark for the efficacy and safety of new disease-modifying therapies. At the dosages used for the treatment of RA, methotrexate has been shown to stimulate adenosine release from cells, producing an anti-inflammatory effect. The clinical efficacy of leflunomide, an inhibitor of pyrimidine synthesis, appears similar to that of methotrexate; it has been shown in well-designed trials to be effective for the treatment of RA as monotherapy or in combination with methotrexate and other DMARDs.
Although similar to the other DMARDs in its slow onset of action, hydroxychloroquine has not been shown to delay radiographic progression of disease and thus is not considered to be a true DMARD. In clinical practice, hydroxychloroquine is generally used for treatment of early, mild disease or as adjunctive therapy in combination with other DMARDs. Sulfasalazine is used in a similar manner and has been shown in randomized, controlled trials to reduce radiographic progression of disease. Minocycline, gold salts, penicillamine, azathioprine, and cyclosporine have all been used for the treatment of RA with varying degrees of success; however, they are used sparingly now due to their inconsistent clinical efficacy or unfavorable toxicity profile.
BIOLOGICALS
Biologic DMARDs have revolutionized the treatment of RA over the past decade (Table 380-2). They are protein therapeutics designed mostly to target cytokines and cell-surface molecules. The TNF inhibitors were the first biologicals approved for the treatment of RA. Anakinra, an IL-1 receptor antagonist, was approved shortly thereafter; however, its benefits have proved to be relatively modest compared with the other biologicals and is rarely used for the treatment of RA with the availability of other more effective agents. Abatacept, rituximab, and tocilizumab are the newest members of this class.
Anti-TNF Agents The development of TNF inhibitors was originally spurred by the experimental finding that TNF is a critical upstream mediator of joint inflammation. Currently, five agents that inhibit TNF-α are approved for the treatment of RA. There are three different anti-TNF monoclonal antibodies. Infliximab is a chimeric (part mouse and human) monoclonal antibody, whereas adalimumab and golimumab are humanized monoclonal antibodies. Certolizumab pegol is a pegylated Fc-free fragment of a humanized monoclonal antibody with binding specificity for TNF-α. Lastly, etanercept is a soluble fusion protein comprising the TNF receptor 2 in covalent linkage with the Fc portion of IgG1. All of the TNF inhibitors have been shown in randomized controlled clinical trials to reduce the signs and symptoms of RA, slow radiographic progression of joint damage, and improve physical function and quality of life. Anti-TNF drugs are typically used in combination with background methotrexate therapy. This combination regimen, which affords maximal benefit in many cases, is often the next step for treatment of patients with an inadequate response to methotrexate therapy. Etanercept, adalimumab, certolizumab pegol, and golimumab have also been approved for use as monotherapy.
Anti-TNF agents should be avoided in patients with active infection or a history of hypersensitivity to these agents and are contraindicated in patients with chronic hepatitis B infection or class III/IV congestive heart failure. The major concern is the increased risk for infection, including serious bacterial infections, opportunistic fungal infection, and reactivation of latent tuberculosis. For this reason, all patients are screened for latent tuberculosis according to national guidelines prior to starting anti-TNF therapy (Chap. 202). In the United States, patients are skin tested using an intradermal injection of purified protein derivative (PPD); individuals with skin reactions of more than 5 mm are presumed to have had previous exposure to tuberculosis and are evaluated for active disease and treated accordingly. The QuantiFERON IFN-γ release assay may also be used in selected circumstances to screen for previous exposure to tuberculosis.
Anakinra Anakinra, the recombinant form of the naturally occurring IL-1 receptor antagonist. Although anakinra has seen limited use for the treatment of RA, it has enjoyed a resurgence of late as an effective therapy of some rare inherited syndromes dependent on IL-1 production, including neonatal-onset inflammatory disease, Muckle-Wells syndrome, and familial cold urticaria, as well as systemic juvenile-onset inflammatory arthritis and adult-onset Still’s disease. Anakinra should not be combined with an anti-TNF drug due to the high rate of serious infections as observed with this regimen in a clinical trial.
Abatacept Abatacept is a soluble fusion protein consisting of the extracellular domain of human CTLA-4 linked to the modified portion of human IgG. It inhibits the co-stimulation of T cells by blocking CD28-CD80/86 interactions and may also inhibit the function of antigen-presenting cells by reverse signaling through CD80 and CD86. Abatacept has been shown in clinical trials to reduce disease activity, slow radiographic progression of damage, and improve functional disability. Many patients receive abatacept in combination with methotrexate or another DMARD such as leflunomide. Abatacept therapy has been associated with an increased risk of infection.
Rituximab Rituximab is a chimeric monoclonal antibody directed against CD20, a cell-surface molecule expressed by most mature B lymphocytes. It works by depleting B cells, which in turn, leads to a reduction in the inflammatory response by unknown mechanisms. These mechanisms may include a reduction in autoantibodies, inhibition of T cell activation, and alteration of cytokine production. Rituximab has been approved for the treatment of refractory RA in combination with methotrexate and has been shown to be more effective for patients with seropositive than seronegative disease. Rituximab therapy has been associated with mild to moderate infusion reactions as well as an increased risk of infection. Notably, there have been isolated reports of a potentially lethal brain disorder, progressive multifocal leukoencephalopathy (PML), in association with rituximab therapy, although the absolute risk of this complication appears to be very low in patients with RA. Most of these cases have occurred on a background of previous or current exposure to other potent immunosuppressive drugs.
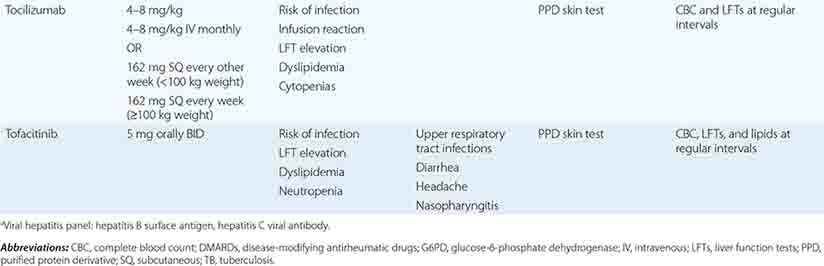
Tocilizumab Tocilizumab is a humanized monoclonal antibody directed against the membrane and soluble forms of the IL-6 receptor. IL-6 is a proinflammatory cytokine implicated in the pathogenesis of RA, with detrimental effects on both joint inflammation and damage. IL-6 binding to its receptor activates intracellular signaling pathways that affect the acute-phase response, cytokine production, and osteoclast activation. Clinical trials attest to the clinical efficacy of tocilizumab therapy for RA, both as monotherapy and in combination with methotrexate and other DMARDs. Tocilizumab has been associated with an increased risk of infection, neutropenia, and thrombocytopenia; however, the hematologic abnormalities appear to be reversible upon stopping the drug. In addition, this agent has been shown to increase LDL cholesterol; however, it is not known as yet if this effect on lipid levels increases the risk for development of atherosclerotic disease.
SMALL-MOLECULE INHIBITORS
Because some patients do not adequately respond to conventional DMARDS or biologic therapy, other therapeutic targets have been investigated to fill this gap. Recently, drug development in RA has focused attention on the intracellular signaling pathways that transduce the positive signals of cytokines and other inflammatory mediators that create the positive feedback loops in the immune response. These synthetic DMARDs aim to provide the same efficacy as biological therapies in an oral formulation.
Tofacitinib Tofacitinib is a small-molecule inhibitor that primarily inhibits JAK1 and JAK3, which mediate signaling of the receptors for the common γ-chain-related cytokines IL-2, -4, -7, -9, -15, and -21 as well as IFN-γ and IL-6. These cytokines all play roles in promoting T and B cell activation as well as inflammation. Tofacitinib, an oral agent, has been shown in randomized, placebo-controlled clinical trials to improve the signs and symptoms of RA significantly over placebo. Major adverse events include elevated serum transaminases indicative of liver injury, neutropenia, increased cholesterol levels, and elevation in serum creatinine. Its use is also associated with an increased risk of infections. Tofacitinib can be used as monotherapy or in combination with methotrexate.
ACR/EULAR PROVISIONAL DEFINITION OF REMISSION IN RHEUMATOID ARTHRITIS |
Source: Adapted from DT Felson et al: Arthritis Rheum 63:573, 2011.
PHYSICAL THERAPY AND ASSISTIVE DEVICES
All patients should receive a prescription for exercise and physical activity. Dynamic strength training, community-based comprehensive physical therapy, and physical-activity coaching (emphasizing 30 min of moderately intensive activity most days a week) have all been shown to improve muscle strength and perceived health status. Foot orthotics for painful valgus deformity decrease foot pain and resulting disability and functional limitations. Judicious use of wrist splints can also decrease pain; however, their benefits may be offset by decreased dexterity and a variable effect on grip strength.
SURGERY
Surgical procedures may improve pain and disability in RA—most notably the hands, wrists, and feet, typically after the failure of medical therapy with varying degrees of reported long-term success. For large joints, such as the knee, hip, shoulder, or elbow, total joint arthroplasty is an option for advanced joint disease. A few surgical options exist for dealing with the smaller hand joints. Silicone implants are the most common prosthetic for MCP arthroplasty and are generally implanted in patients with severe decreased arc of motion, marked flexion contractures, MCP joint pain with radiographic abnormalities, and severe ulnar drift. Arthrodesis and total wrist arthroplasty are reserved for patients with severe disease who have substantial pain and functional impairment. These two procedures appear to have equal efficacy in terms of pain control and patient satisfaction. Numerous surgical options exist for correction of hallux valgus in the forefoot, including arthrodesis and arthroplasty, as well as primarily arthrodesis for refractory hindfoot pain.
OTHER MANAGEMENT CONSIDERATIONS
Pregnancy Up to 75% of female RA patients will note overall improvement in symptoms during pregnancy, but often will flare after delivery. Flares during pregnancy are generally treated with low doses of prednisone; hydroxychloroquine and sulfasalazine are probably the safest DMARDs to use during pregnancy. Methotrexate and leflunomide therapy are contraindicated during pregnancy due to their teratogenicity in animals and humans. The experience with biologic agents has been insufficient to make specific recommendations for their use during pregnancy. Most rheumatologists avoid their use in this setting; however, exceptions are considered depending on the circumstances.
Elderly Patients RA presents in up to one-third of patients after the age of 60; however, older individuals may receive less aggressive treatment due to concerns about increased risks of drug toxicity. Studies suggest that conventional DMARDs and biologic agents are equally effective and safe in younger and older patients. Due to comorbidities, many elderly patients have an increased risk of infection. Aging also leads to a gradual decline in renal function that may raise the risk for side effects from NSAIDs and some DMARDS, such as methotrexate. Renal function must be taken into consideration before prescribing methotrexate, which is mostly cleared by the kidneys. To reduce the risks of side effects, methotrexate doses may need to be adjusted downward for the drop in renal function that usually comes with the seventh and eighth decades of life. Methotrexate is usually not prescribed for patients with a serum creatinine greater than 2 mg/dL.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


