Surgical Repair of Tracheoesophageal Fistula and Esophageal Atresia
Scott A. Engum
Frederick Rescorla
Esophageal atresia (EA) and tracheoesophageal fistula (TEF) are common neonatal conditions that remain a significant challenge to children’s surgical specialists. The first description of EA is attributed to William Durston in 1670. In 1696, Thomas Gibson described the association of EA with a distal TEF. In 1939, Thomas Lanman in Boston and Logan Leven of Minneapolis reported the successful management of babies who had EA using a staged repair that initially created a gastrostomy followed by ligation of the fistula and subsequent esophageal replacement using an antethoracic skin tube. The first successful primary repair was performed by Cameron Haight of Ann Arbor, Michigan, in 1941. Since then, continuous refinements in clinical management have occurred associated with improved surgical technique, advances in neonatal anesthesia, development of infant ventilators, and modern sophisticated neonatal intensive care. These advances have allowed survival of even low-birth-weight infants, many of whom have associated anomalies that formerly resulted in death.
Whereas EA with TEF is a relatively common foregut anomaly, the embryologic events leading to this congenital malformation remain poorly understood. Although
there is no strong support for an inheritable mechanism, the pathogenesis of congenital EA/TEF is likely multifactorial. A slightly increased incidence of EA/TEF occurs in twins, but most cases occur sporadically. Chromosomal anomalies have been reported in up to 10% of cases, but no specific chromosomal defect has been confirmed as an etiological factor. However, four genes, MYCN, CHD7, MID1, and SOX2, have been implicated in rare cases of syndromic EA/TEF. Knockout studies in mice have elucidated the functions of different genes involved in foregut development, implicating genes like Sonic hedgehog (SHH), Gli2, Gli3, Foxf1, and TTF-1 in the development of EA/TEF. However, other than a rare case report of a deletion of SHH, no mutations in these genes have been reported in humans with EA or TEF. Environmental teratogens such as oral contraceptives, methimazole, and benectin have also been implicated in the pathogenesis of EA/TEF and the VACTERL association, but in the majority of patients no specific teratogen has been found. Insight into the pathogenesis of EA/TEF and the VACTERL association has been gained using the teratogen model of adriamycin-induced EA/TEF, and investigation of fistula tissue for markers of respiratory versus foregut origin. Further characterization of the molecular makeup of the human neonatal fistula tract may provide insight into the precise embryogenesis of this anomaly and its postoperative outcome.
there is no strong support for an inheritable mechanism, the pathogenesis of congenital EA/TEF is likely multifactorial. A slightly increased incidence of EA/TEF occurs in twins, but most cases occur sporadically. Chromosomal anomalies have been reported in up to 10% of cases, but no specific chromosomal defect has been confirmed as an etiological factor. However, four genes, MYCN, CHD7, MID1, and SOX2, have been implicated in rare cases of syndromic EA/TEF. Knockout studies in mice have elucidated the functions of different genes involved in foregut development, implicating genes like Sonic hedgehog (SHH), Gli2, Gli3, Foxf1, and TTF-1 in the development of EA/TEF. However, other than a rare case report of a deletion of SHH, no mutations in these genes have been reported in humans with EA or TEF. Environmental teratogens such as oral contraceptives, methimazole, and benectin have also been implicated in the pathogenesis of EA/TEF and the VACTERL association, but in the majority of patients no specific teratogen has been found. Insight into the pathogenesis of EA/TEF and the VACTERL association has been gained using the teratogen model of adriamycin-induced EA/TEF, and investigation of fistula tissue for markers of respiratory versus foregut origin. Further characterization of the molecular makeup of the human neonatal fistula tract may provide insight into the precise embryogenesis of this anomaly and its postoperative outcome.
EA and TEF are relatively common congenital anomalies with an incidence of one in 3,000 to 4,000 live births. Antenatal detection of EA/TEF is challenging. Definitive prenatal diagnosis may be limited by clinical and technical variations. The physician may suspect an EA/TEF on the basis of a third trimester ultrasound demonstrating polyhydramnios with or without a stomach bubble. Some have visualized the upper neck pouch suspecting a blind-ending esophagus. Despite various markers, antenatal detection rates have been previously reported in only 10% to 42% of cases. In addition, the prognostic value of prenatal diagnosis of EA/TEF is unclear. Early identification may allow counseling by a pediatric surgeon and prepare the family emotionally; however, some have shown that prenatal detection neither affects neonatal outcome nor identifies a group at increased risk for neonatal morbidity and mortality. The authors showed favorable outcomes with or without prenatal suspicion. This may reflect the comprehensive care readily available at a tertiary care facility.
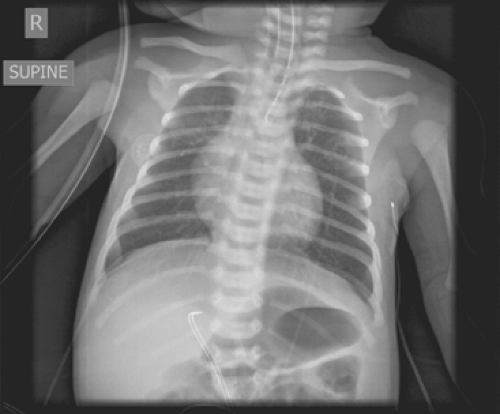 Fig. 1. Radiograph demonstrating a blind-ending proximal esophageal pouch with feeding tube and presence of gastric distention. |
Common findings on presentation include excessive salivation due to pooling of secretions in the proximal atretic esophageal pouch, respiratory distress, and cyanosis due to aspiration. Abdominal distention may occur as a result of inspired air passing through the fistula into the stomach, causing gastric dilatation (Fig. 1) with subsequent reflux of gastric juice upward through the fistula and into the lungs, resulting in pneumonitis.
Some babies have severe respiratory distress and require mechanical ventilation before their admission to a specialized high-risk neonatal center. An evaluation of infants with EA, respiratory distress, and a clear chest radiograph concluded that the respiratory distress is related to both tracheomalacia and upper-airway obstruction that may cause miliary atelectasis not detected by a conventional radiograph with intrapulmonary shunting and hypoxemia.
Associated congenital anomalies are common, occurring in 146 of 227 cases (65%) treated in our facility; congenital heart defects are the most common defect, noted in 38% of cases. Musculoskeletal anomalies were seen in 19% of cases, central nervous system abnormalities in 15%, renal anomalies in 15%, and associated abnormalities of the alimentary tract (including duodenal atresia, malrotation, and imperforate anus) in 13%.
There are five major anatomic variants of EA (Fig. 2). Type A, atresia without a fistula, occurs in approximately 5% to 13% of cases; type B, in which a proximal atresia and fistula from the proximal esophagus are present, occurs in 1% of cases; and the most common variant, type C, with proximal EA and a distal TEF, occurs in 78% to 86% of cases. We had five type D patients (2%) who had a proximal EA and a double fistula (both proximal and distal); an H-type TEF may occur in the absence of EA (type E) in 2% to 6% of cases. For the purpose of this chapter, we describe the operative management of the most common variants, beginning with type C and also discussing types A and E.
Clinical diagnosis of EA/TEF is confirmed by passing an oral gastric tube into the stomach. The tube typically stops at the 12-cm mark and a chest radiograph should be obtained to confirm tube location. In the typical EA/TEF one can see the oral gastric tube either coiled in the upper esophageal pouch or coming to an abrupt end (Fig. 3). During radiographic evaluation, the surgeon will note the absence or presence of air in the gastrointestinal tract. Absence of air raises concern for a type A or B variant. One would assume that with the presence of a patent fistula, air would appear in the stomach even with the first breath. However, a recent report has shown that the absence of gas in the stomach on X-ray, undertaken within the first 4 hours of life, does not necessarily mean the absence of a distal fistula. Data suggest that it may be necessary to acquire a second X-ray at least 4 hours before the child is taken for corrective surgery. The presence of air in the stomach usually confirms the presence of a distal fistula in type C or D variants. Contrast studies are mostly unnecessary to make the appropriate diagnosis and may be troublesome. Aspiration of radiographic contrast material in the tracheobronchial tree may
cause serious morbidity. If needed, contrast studies are best undertaken in centers where appropriate experience and facilities for neonatal emergency resuscitation and ventilation are available. Bedside flexible bronchoscopy via the endotracheal tube can be initiated if anatomy clarification is necessary prior to the operating room.
cause serious morbidity. If needed, contrast studies are best undertaken in centers where appropriate experience and facilities for neonatal emergency resuscitation and ventilation are available. Bedside flexible bronchoscopy via the endotracheal tube can be initiated if anatomy clarification is necessary prior to the operating room.
Table 1 Waterston Classification | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
The timing of operation must be individualized according to the infant’s clinical condition. Although the Waterston classification system, devised in 1962 to categorize babies with EA, was used extensively in the past, we no longer use these guidelines and individualize patients according to their clinical status. Assessment of the physiologic status of neonates has refined the Waterston criteria; birth weight, gestational age, and pulmonary status, previously considered essential determinants, are no longer the prime considerations.
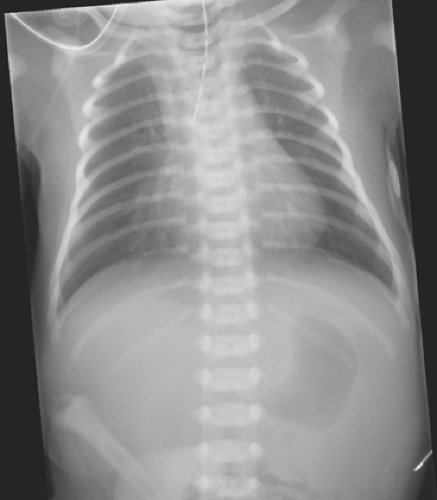 Fig. 3. Radiograph demonstrating orogastric tube in the proximal atretic pouch and air below the diaphragm. |
Although some investigators question the validity of Waterston’s classification (Table 1), Dunn and colleagues have shown that it continues to be useful if one simply combines Groups A and B into a single risk status. This stratification reliably selects infants at risk of dying, and allows the results to be compared from center to center. Newer proposals have limited information available; however, all centers have information available with regard to the Waterston criteria. Some believe this classification is still valid, useful, and preserves the continuity of data comparison in the literature.
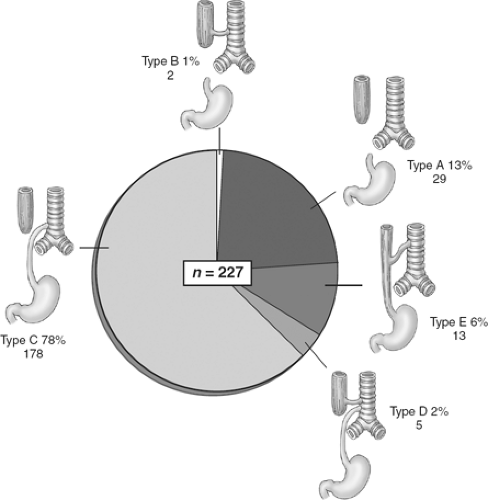 Fig. 2. Classification of esophageal atresia and tracheoesophageal fistula. |
The Montreal classification recommended by Poenaru is a method designed for classifying infants with EA and reflects both the significant progress in neonatal care that has been achieved during the past three decades and the severity of associated congenital anomalies and pulmonary disease observed. This classification system (Table 2) proposes that birth weight is not a significant factor for mortality, but the need for preoperative ventilation is. Low birth weight and prematurity alone do not dictate a worse prognosis for infants with EA, TEF, or both, and therapy is more individualized. The Montreal prognostic criteria identify infants that are dependent on ventilator
support preoperatively and have life-threatening anomalies and are relatively simple and easy to define in a prospective fashion.
support preoperatively and have life-threatening anomalies and are relatively simple and easy to define in a prospective fashion.
Table 2 Montreal Classification | ||||||
|---|---|---|---|---|---|---|
| ||||||
Spitz proposed that birth weight (<1,500 g) and major congenital heart disease (associated with cyanosis and/or heart failure requiring surgery) are the major factors associated with mortality (Table 3). Encinas et al. showed that with only a few exceptions, the diagnosis of congenital heart disease does not change the timing and the modalities of EA repair. In addition, the presence of a right aortic arch (RAA), aberrant right subclavian artery, or dextrocardia does not change the chances of a successful esophageal anastomosis by the usual approach. Yagyu has proposed the Bremen classification, which modifies Spitz’s classification to include preoperative pulmonary status. There are few published reports comparing these classification systems, however; Sinha et al. evaluated four prognostic criteria (Spitz, Waterston, Bremen, and Montreal) and showed identical results for Waterston class A and B as well as for the Bremen “without complications” group. Furthermore, there was no statistically significant difference between Spitz type I and type II. There was, however, a significant variation after combining the results for Spitz type I and II and comparing them to type III. On the basis of these results, the authors recommend further modified prognostic criteria for infants with EA/TEF where group A would include infants with either a single poor prognostic risk factor (i.e., weight below 1.5 kg or a major cardiac anomaly) or isolated EA/TEF where the prognosis should be excellent (survival 98%). The alternate group B would include infants affected by both negative risk factors and EA/TEF and have a poor prognosis (survival 33%). Konkin in a retrospective review suggested the Spitz classification system was the most reliable in predicting prognosis for patients with EA/TEF. The mortality rate for EA/TEF remains on the decline and the most important direction in the management focuses on reducing the morbidity related to associated anomalies, especially major cardiac defects. As continued improvement in neonatal critical care management occurs, classification systems will need to expand and redefine themselves.
Table 3 Spitz Classification | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
In general, whatever classification system is used, infants that have stable cardiac and respiratory status undergo expeditious chest exploration and repair. Anomalies that are not life-threatening in the presence of physiologic derangements that are easily correctable are included in the determination of stability. Intercostal retractions, oxygen requirements, endotracheal intubation, and minor pulmonary infiltrates are not considered absolute contraindications to immediate repair as long as the infant is stable. Smaller infants, even those with minimal respiratory compromise and associated anomalies after appropriate stabilization, undergo similar repair within 24 hours. Poor-risk infants, especially those that are premature (less than 1,000 grams) and have severe respiratory ailments or congestive heart failure (or both), may require some delay in therapy and may initially be treated with an emergency gastrostomy along with distal esophageal obstruction whether by a Fogarty balloon obstruction (Fig. 4) or distal esophageal vessel loop occlusion, continuous proximal atretic pouch suction, and administration of intravenous antibiotics. In selective cases, a proximal Fogarty balloon can be placed via the trachea with occlusion of the esophageal fistula. Occasionally, urgent thoracotomy with ligation and division of the TEF, followed by delayed esophageal repair, may be necessary when respiratory support is required and inspired air is lost via the fistula. Definitive esophageal repair is attempted when the infant’s condition stabilizes.
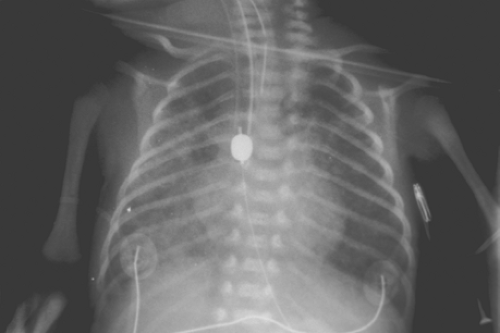 Fig. 4. Radiograph illustrating a Fogarty balloon catheter in the distal esophagus that has been passed cephalad via the gastrostomy site to occlude the distal esophagus. |
The classical method for surgical management of EA/TEF is closure of fistula and primary anastomosis of proximal and distal end through a right thoracotomy. However, the RAA in association with EA/TEF is challenging for primary repair. The most appropriate surgical technique remains controversial. The incidence of RAA has been suggested 1/1,000 in the general population. Previous reports estimate the RAA is associated with 5% of EA/TEF patients, and Bicakci et al. showed the incidence as high as 13%. Evaluation of associated anomalies in newborns with EA/TEF is necessary to improve the outcome of surgical repair and to predict the prognosis. Echocardiography is a noninvasive technique for the diagnosis of congenital heart disease; however, the possibility of defining RAA with echocardiography is low in patients with EA/TEF. Some believe the dissection of a fistula and anastomosis is too complicated in a case of RAA leading to change of the chest exploration side in these patients; however, Bicakci and colleagues
showed they could achieve primary repair in all patients with RAA through a right-sided thoracotomy. Although some believe that echocardiographic evaluation for RAA is not beneficial, many surgeons continue to acquire this study not only to evaluate complex congenital heart disease, but to guide them in a possible surgical chest exploration choice. Further diagnostic evaluation of a patient with an aortic arch that cannot be easily visualized should be considered before proceeding with definitive repair in the face of a possible vascular ring.
showed they could achieve primary repair in all patients with RAA through a right-sided thoracotomy. Although some believe that echocardiographic evaluation for RAA is not beneficial, many surgeons continue to acquire this study not only to evaluate complex congenital heart disease, but to guide them in a possible surgical chest exploration choice. Further diagnostic evaluation of a patient with an aortic arch that cannot be easily visualized should be considered before proceeding with definitive repair in the face of a possible vascular ring.
Esophageal repair is best carried out in the operating room rather than in the newborn unit. This allows all individuals involved in the surgical care of the infant to be in a zone of technical comfort. The room temperature is adjustable and is kept at approximately 80ºF to maintain thermal neutrality. The procedure may be performed under an overhead warmer in an attempt to maintain the baby’s temperature if room adjustments are not obtainable. The baby is placed on a warming blanket and carefully monitored with an arterial catheter (umbilical or peripheral) in place, an electrocardiogram monitor, a pulse oximeter, and a thermal probe. The baby’s legs are wrapped in swaddling cloth, and a cap is placed on the head to retain body heat.
An awake intubation is commonly performed to allow spontaneous breathing to occur, and the endotracheal tube is positioned with the bevel facing posteriorly to occlude the fistula. The goal of airway management in a patient with EA/TEF is to maintain adequate ventilation of the lungs without ventilating the fistula, which can lead to ineffective pulmonary ventilation, gastric distention or rupture, hypotension, or reflux of gastric contents. There are two major strategies to achieve this goal: proper positioning of an endotracheal tube or catheter occlusion of the TEF, both of which have potential difficulties. Miyamoto and colleagues have utilized a bifurcated tracheal tube for a neonate with a TEF. Tercan described one-lung ventilation of a preterm newborn accomplished by left mainstem intubation during the repair. There are advantages and disadvantages to these methods and further refinement will be required prior to widespread utilization. A small, flexible fiberoptic bronchoscope is passed down the endotracheal tube to ascertain the location of the end of the tube and the location of the TEF opening. An occasional fistula originating from the proximal pouch can also be noted during endoscopy. Some pediatric surgeons routinely perform endoscopy of the proximal pouch in order to identify the presence of a proximal fistula.
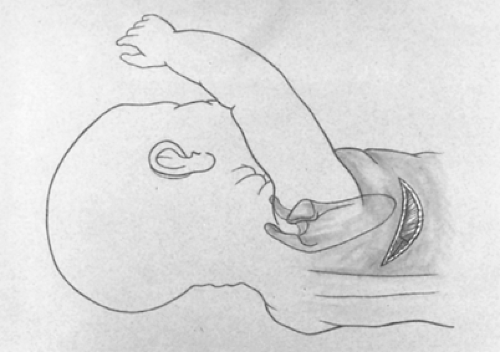 Fig. 5. A posterolateral incision is made just below the tip of the scapula. The latissimus dorsi muscle is divided while the serratus anterior muscle is retracted anteriorly and medially. |
The patient is positioned left side down for a right chest exploration. A folded towel or soft rubber support is placed under the left axilla to ensure that the left chest can expand adequately. The baby’s chest is prepared with a warm iodophor solution and draped appropriately. A 10 French Replogle tube is placed in the proximal esophageal pouch. If a previous gastrostomy was performed, it is opened and placed on straight drainage to avoid gastric distention during the procedure.
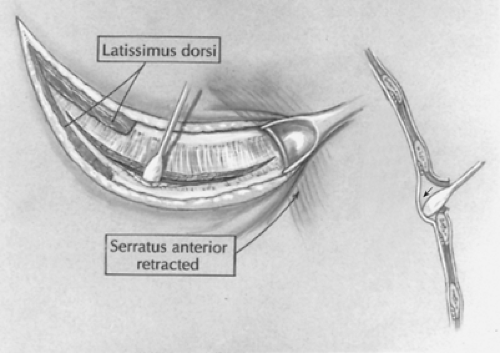 Fig. 6. The fourth intercostal space is identified and entered, and an extrapleural dissection is carried out using a moistened cotton-tipped applicator (arrow). |
Open Operative Technique
A short posterolateral incision is made below the level of the tip of the scapula (Fig. 5), and hemostasis is obtained with electrocoagulation. The latissimus dorsi muscle is identified and retracted posteriorly, but occasionally divided (Fig. 6). The auscultatory space of Korotkoff is identified, and the scapula is retracted superiorly and the serratus anterior muscle medially. It is usually unnecessary to divide the serratus muscle, thus avoiding the risk of developing a winged scapula. The fourth intercostal space is identified and entered. The intercostal muscles are carefully divided to avoid entering the pleura. An extrapleural dissection is carried out using either a small periosteal elevator or a moistened cotton-tipped applicator. An extrapleural plane is established, and an infant rib spreader is inserted to improve exposure. The intact pleura is dissected superiorly to the thoracic inlet and posteriorly to identify the azygos vein. The intact pleura and underlying lung are retracted medially with moistened sponges under a soft malleable retractor. The dissection continues with the use of
a moist peanut sponge until the azygos is fully mobilized and the mediastinal pleura over the vein is opened (Fig. 7). The azygos vein is divided between two 4-0 suture ties. This vein is often the marker for the site of the TEF as it enters the trachea. A branch of the vagus nerve usually passes over the area as well.
a moist peanut sponge until the azygos is fully mobilized and the mediastinal pleura over the vein is opened (Fig. 7). The azygos vein is divided between two 4-0 suture ties. This vein is often the marker for the site of the TEF as it enters the trachea. A branch of the vagus nerve usually passes over the area as well.
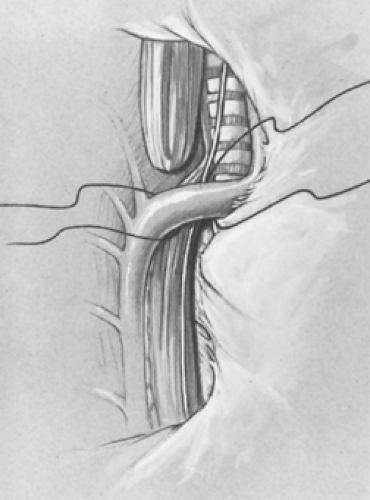 Fig. 7. The intact pleura is retracted. The azygos vein is isolated, divided, and retracted anteriorly with the pleura, allowing identification of the tracheoesophageal fistula. |
After ligation and division of the vein, the TEF is identified and carefully dissected free near the trachea (Fig. 8A). It is important to avoid dividing small vessels to the midesophagus that arise directly from the aorta. A vascular loop is placed around the TEF, and a fine 5-0 absorbable suture on a small tapered needle is placed at the end of the TEF on the esophageal side to avoid tracheal narrowing by the closure (Fig. 8B). The TEF is divided using fine tenotomy scissors, and the tracheal side is oversewn (Fig. 8C) with either a running or interrupted 5-0 suture. The suture line is then placed under saline, and the anesthesiologist is requested to slightly hyperinflate the lungs to ensure that the tracheal closure is airtight. Mediastinal pleura is used to cover the tracheal suture line to reduce the risk of a recurrent TEF should an anastomotic leak occur postoperatively. Some surgeons have utilized an intercostals muscle flap, pericardium, the azygous vein, or autologous tissue products to maintain separation of the two suture lines.
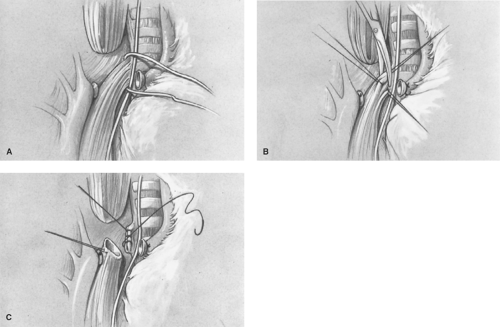 Fig. 8. A: The tracheoesophageal fistula site is mobilized circumferentially distal to the fistula, and the area is encircled with a vessel loop. B: Traction sutures are placed, with great care taken not to encroach on the lumen of the trachea, which can result in narrowing with closure. Division of the fistula is then performed using tenotomy scissors. C: The tracheal end is closed with a 5-0 continuous or interrupted suture. The closure is tested with positive-pressure ventilation to rule out a leak. The tracheal suture line is covered with mediastinal pleura to reduce the risk of recurrent tracheoesophageal fistula should an anastomotic leak occur. |
The upper atretic esophageal pouch is identified with the aid of the anesthesiologist, who pushes down on the end of the pouch with a previously placed oral tube. Placing a traction suture through the tip of the pouch and the oral tube (Fig. 9) facilitates the dissection of the proximal pouch. The atretic esophagus is mobilized up to the level of the thoracic inlet and into the neck to acquire adequate length to achieve primary repair by constructing an anastomosis without tension. The rich blood supply to the more hypertrophic upper pouch is derived from the inferior thyroid artery. Clearing the esophagus from the mediastinal tissues can be done by blunt dissection, using a 45-degree clamp and a fine-tipped electrocoagulator. Dissection of the proximal atretic pouch from the tracheal wall can be difficult and requires meticulous attention and gentle technique to avoid injury of or entry into the trachea. Rarely (in 2% of cases), a type D variant (double fistula) is encountered, with the second fistula located in the upper pouch. Traction sutures are placed at the 3 and 9 o’clock positions on the open distal esophagus, and by placing gentle traction in opposite directions, the proximity of the upper and lower esophageal ends is determined. Extensive distal esophageal mobilization should be avoided to reduce the risk of ischemia to the midesophagus caused by damaging its limited blood supply from small arterial braches that come directly from the aorta. The major blood supply to the midesophagus in these cases comes through the TEF, which has already been divided. If the distal
rim of the esophageal tissue is cyanotic, it should be carefully debrided. The distal esophageal opening is gently dilated with a right-angle clamp. Full-thickness lateral sutures of 4-0 suture are placed in the distal esophagus. Each suture must include a healthy full-thickness bite, including mucosa and the smooth muscle of the esophageal wall. The needles should be left in place so that the esophageal wall can be sutured to the upper pouch.
rim of the esophageal tissue is cyanotic, it should be carefully debrided. The distal esophageal opening is gently dilated with a right-angle clamp. Full-thickness lateral sutures of 4-0 suture are placed in the distal esophagus. Each suture must include a healthy full-thickness bite, including mucosa and the smooth muscle of the esophageal wall. The needles should be left in place so that the esophageal wall can be sutured to the upper pouch.
The distal portion of the larger, thicker proximal atretic pouch is incised directly over the tube located in the pouch lumen to determine its dependent area. Entry can be accomplished with a scalpel, tenotomy scissors, or electrocautery. The size of the opening should be similar to or just slightly larger than the distal esophagus after it has been dilated. A full-thickness suture bite is placed in the midportion of the posterior wall of both ends of the esophagus, with the knots tied on the outside (Fig. 10A). Using this suture and the previously placed corner sutures for traction, the one-layer posterior suture line is completed with a few additional interrupted sutures. We typically place all the sutures before tying them to distribute the tension evenly along the suture line. Some surgeons prefer to place the posterior suture line with the knots within the lumen. A nasogastric tube (No. 6 to 10 French) is then passed from above by the anesthesiologist through the anastomosis into the stomach. The anterior wall of the esophageal anastomosis is closed with full-thickness, interrupted sutures (Fig. 10B and C), completing the single-layer repair. Some surgeons find it useful to place a small catheter in the upper esophagus above the suture line, and carefully instill 5 to 10 mL of saline to test the anastomotic suture line for a leak.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


