Surgical Anatomy of the Diaphragm
Richard R. Ricketts
Marios Loukas
Lee J. Skandalakis
David A. McClusky III
Petros Mirilas
Embryogenesis
Embryogenesis of the diaphragm comes from the following sources: transverse septum (ventrally), paired pleuroperitoneal membranes (posteriorly), skeletal muscle derived in situ from the body wall, and migration of cervical myotomes (peripherally) (Fig. 1). Some authors of the previous decades believed that the mesentery of the esophagus also contributed to the diaphragm.
Transverse Septum
The septum transversum is a mesenchymal bridge arising from the coelomic wall located between the pericardial cavity and the umbilical vesicle (Fig. 2). In early stages this mesenchyme mass separates ventrally the pleural and peritoneal cavities. It extends from the ventral body wall to the ventral border of the foregut.
The cranial part of the septum contributes to the connective tissue of the pericardium and is finally covered by pericardium and pleura. The ventral part of the septum constitutes a sagittal mesentery in which the liver develops. The septum transversum, which originates at a cervical level, gives genesis to the major part of the diaphragm (central tendon and ventral part). Therefore, the major innervation of the diaphragm is supplied from the phrenic nerve, derived from C3, C4, and C5.
Pleuroperitoneal Membranes
The pleuroperitoneal membranes close the right and left communication between the pleural and peritoneal cavities at approximately the 8th embryonic week. The pleuroperitoneal canals are closed by fusion of their edges. Growth of the adjacent adrenals brings the closing edges from posterolaterally to a more anteromedial final location. The right canal closes earlier. Originally, the pleuroperitoneal membranes form a large part of the developing diaphragm, but relative growth of other elements reduces their contribution to a small area.
Muscles of the Body Wall
Myotomes of the 7th to 12th segments contribute the lateral component of the diaphragm by caudal excavation of the thoracic wall to form the costodiaphragmatic recesses. This process produces the final domed shape of the diaphragm, and explains innervation of the peripheral part of the diaphragm by the lower intercostals nerves.
Except from the in situ muscles of the body wall, cervical myotomes accompany the phrenic nerve, migrate, and contribute to the skeletal muscle of the diaphragm. This explains the innervation of the central part of the diaphragm by the phrenic nerve.
Descent of the Diaphragm
In the 3rd week, the transverse septum lies at the level of C3, and the developing diaphragm descends to its final position at the level of L1 by the 8th week (Fig. 3). The phrenic nerve, which originates from the third to fifth cervical levels, is carried caudad with the descending diaphragm.
Diaphragmatic Anomalies
A number of areas of the diaphragm can give way under pressure from the abdominal viscera. Most diaphragmatic hernias start in these small areas of weakness and enlarge with age. The specific hernias are described in the following sections and summarized in Table 1.
Congenital Anomalies
Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernias (CDHs) usually occur through the posterolateral foramen of Bochdalek and rarely through the substernal foramen of Morgagni. They are more frequent on the left side (80%) and are very rarely bilateral. CDH is accompanied by pulmonary hypoplasia (both lungs) and pulmonary hypertension.
Many theories have been proposed to explain the appearance of posterolateral diaphragmatic defects:
Improper development of the pleuroperitoneal membrane
Failure of muscularization of the lumbocostal trigone resulting in an area of weakness
Premature return of the intestines into the abdominal cavity when the pleuroperitoneal canal is still “open”
Abnormal persistence of lung in the pleuroperitoneal canal
Abnormal development of the early lung and posthepatic membrane
Abnormal muscle innervation by the phrenic nerve
The most popular theory is that the lung hypoplasia is secondary and a direct consequence of a primary diaphragmatic defect: inhibited or delayed closure of the pleuroperitoneal canal by 8 weeks’ gestation allows the fetal intestine, returning from the extracoelomic cavity at 8 to 10 weeks, to enter the thoracic cavity and impair lung growth and development. However, this turn of events has never been observed in embryos or fetuses. Another popular theory is that the lung is primarily hypoplastic and that the hypoplastic lung in CDH is not the result but the cause of the diaphragmatic defect.
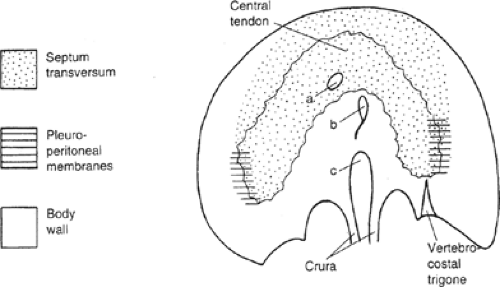 Fig. 1. Scheme showing the probable origin of the diaphragm. Sharp boundaries are not justified. The vertebrocostal trigone does not mark the closure of the pleuroperitoneal canal. a, caval foramen; b, esophageal hiatus; c, aortic hiatus. Based on findings of Wells. (From O’Rahilly R, Müller F. Human Embryology and Teratology, 3rd ed. New York: Wiley-Liss, 2001; with permission.) |
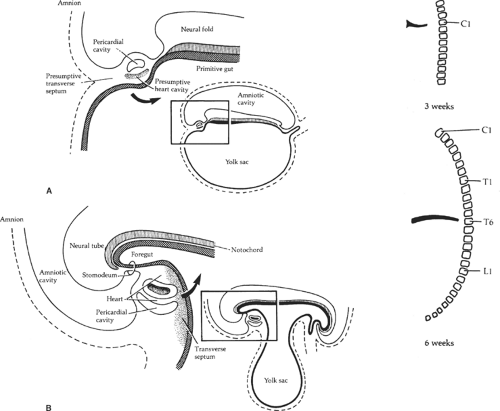 Fig. 2. Formation of the transverse septum. A: The heart and pericardium from anterior to the head of the embryo in the 3rd week. B: Rapid growth of the head rotates the heart and the mesoderm, which become the transverse septum, in the direction indicated by the arrows in the 4th week. |
Using the experimental murine nitrofen model (pregnant rats are given 100 mg of nitrofen orally; fetuses are examined at various points of development and compared with normal ones), Kluth found that the typical features of CDH and pulmonary hypoplasia cannot be explained by inhibited closure of the pleuroperitoneal canals, since these are never large enough to allow for herniation of the gut into the thorax. Also, the maldevelopment of the diaphragm begins early in the embryonic period (corresponding to the 5th week of gestation in humans), and the lungs of the nitrofen-fed rats are already significantly smaller than those of the controls by the end of the embryonic period (8 weeks in humans). Kluth believes that the hypoplastic lung is secondary to the defect in the diaphragm and is caused by ingrowths of liver (not bowel herniation) into the thorax.
Caution must be exercised in extrapolating data from murine nitrofen studies to humans because of the significant anatomical differences between this model and humans: In murine models CDH occurs more often on the right side; the posteromedial diaphragm is affected rather than the posterolateral area as in humans; murine CDHs contain liver (infrequent in humans), and small bowel loops appear late, that is, too late to interfere with lung development; the intrathoracic murine liver appears to be an extra lobe, with the normal liver remaining in the abdominal cavity, rather than a herniation of normal liver into the thorax as is seen in humans.
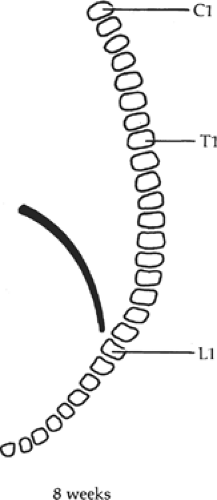 Fig. 3. The descent of the diaphragm during development. The phrenic nerve arises from the third to the fifth cervical segments and follows the diaphragm down to its final position. |
The fetal lamb model is based on the concept that pulmonary abnormalities (pulmonary hypoplasia and pulmonary hypertension) are the result of impaired lung growth secondary to the mass effect of the
herniated viscera. This led to experimental and then clinical trials to reduce the viscera and close the diaphragmatic defect prenatally, in order to allow space for the lung to grow and develop during the rest of gestation. This therapy was offered only to patients with 100% predicted mortality. A randomized trial of open fetal repair versus advanced postnatal care showed that prenatal repair was unsuccessful in the most severe cases and did not reduce mortality in the less severe cases.
herniated viscera. This led to experimental and then clinical trials to reduce the viscera and close the diaphragmatic defect prenatally, in order to allow space for the lung to grow and develop during the rest of gestation. This therapy was offered only to patients with 100% predicted mortality. A randomized trial of open fetal repair versus advanced postnatal care showed that prenatal repair was unsuccessful in the most severe cases and did not reduce mortality in the less severe cases.
Table 1 Characteristics of Diaphragmatic Hernias | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In the 1990s, it was observed that in congenital high airway obstruction syndrome (CHAOS) lung growth is accelerated and the lung becomes hyperplastic, because of the inability of the fetal lung fluid to exit the lung because of tracheal occlusion. In the fetal lamb CDH model, occlusion of the trachea resulted in reduction of the herniated viscera, increased lung volume, and improvement in gas exchange and compliance. Open fetal tracheal occlusion was first performed in a human in 1996. Technology advanced to the point that endoscopic occlusion through an intact uterus (in an attempt to prevent preterm delivery) became feasible by 1999 (“fetendo”). Initially a clip was applied to the trachea. Because of tracheal damage, current technology involves placing a detachable balloon inside the trachea endoscopically. A National Institutes of Health (NIH)–sponsored randomized trial of fetendo balloon occlusion versus postnatal care was started in 2003. After 24 patients were randomized, the study was stopped because there was no survival advantage to the fetendo group versus the controls (73% vs. 77%), and the experimental group experienced more premature rupture of membranes and preterm deliveries than the controls.
Hiatal Hernia
Ascent of the stomach into the thorax through the esophageal hiatus of the diaphragm is a common and poorly understood lesion. It has been found in stillborn infants. O’Rahilly pointed out that the anomaly may lie in a delay of the elongation of the esophagus. Thus the stomach is found in an abnormally high position and the hiatus is formed around the stomach instead of the abdominal esophagus. A congenital hiatal hernia predisposes to an adult hiatal hernia, especially with obesity, as fat accumulates in the mesentery of the esophagus.
As in the adult hiatal hernia, congenital hiatal hernia can be of two types: sliding and paraesophageal (Fig. 4). In sliding hernia the abdominal esophagus and the stomach move in the thorax. The two requirements for a sliding hiatal hernia appear to be an enlarged hiatus and a weakened phrenicoesophageal ligament. Because both conditions are exacerbated by the hernia, the opening is further dilated and the ligament further stretched.
A congenital paraesophageal hernia permits herniation of the fundus of the stomach into the thorax through the hiatus, usually anterior or sometimes besides the esophagus (Fig. 4B). There is a peritoneal sac anterior to the esophagus containing the stomach and, in extreme instances, transverse colon and omentum. Obstruction of the distal esophagus or the stomach is the usual result. The two types of hernia can be combined: a paraesophageal hernia may be associated with a sliding hernia.
Congenital short esophagus can simulate hiatal hernia. It is present in children, although it might be asymptomatic. An overall incidence of 1.53% has been documented. The phrenicoesophageal ligament is normal, there is no hernial sac, and the left gastric artery is not displaced upward. The condition is often familial, and it is more common in males. It is sometimes associated with pyloric stenosis, malrotation, and Marfan syndrome. The authors of this chapter believe that, although it is rare, a short esophagus is a true congenital malformation.
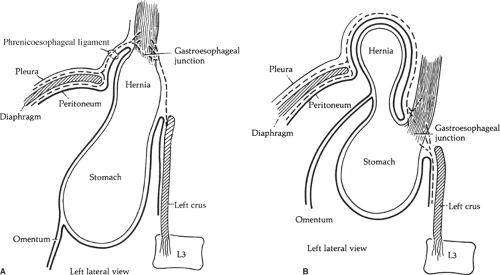 Fig. 4. Hiatal hernias. A: Sliding hiatal hernia seen from the left. The gastroesophageal junction is in the thorax. B: Paraesophageal hernia seen from the left. The gastroesophageal junction is in its normal location; the fundus has herniated into the thorax through the hiatus anterior to the esophagus. |
There are controversies regarding the existence and incidence of short esophagus. Nyhus has stated that a short esophagus is
not congenital, and that the shortening is caused by secondary factors in an esophagus of normal length. He commented that infants who have chalasia develop peptic esophagitis and then shortening of the esophagus.
not congenital, and that the shortening is caused by secondary factors in an esophagus of normal length. He commented that infants who have chalasia develop peptic esophagitis and then shortening of the esophagus.
Barrett’s esophagus must be distinguished from the congenital short esophagus. Barrett’s esophagus is associated with reflux, columnar epithelium, and the stricture cephalad to the gastroesophageal junction. In the congenital esophagus, the distal esophagus is practically a gastric tube, which radiographically cannot be differentiated from hiatal hernia. In this case reflux is significant.
Two conditions must be considered:
Irreducible partially supradiaphragmatic true stomach. The stomach has herniated into the thorax through an enlarged diaphragmatic esophageal hiatus and become fixed. This condition is true fixed hiatal hernia. Occasionally, the fixed hernia is not congenital but acquired, with tissue adhesions.
Partially supradiaphragmatic true stomach existing from birth and not reducible. This condition is true congenital short esophagus and is very rare.
The congenital short esophagus is not associated with a hernial sac. Branches of the left gastric artery do not pass upward through the hiatus. According to Gray and Skandalakis, only a small percentage of hiatal hernias belong to this group. Others have stated that an acquired short esophagus is the result of gastroesophageal reflux of gastric and biliopancreatic fluids.
Horvath et al. provided an interesting commentary on the term “short esophagus”:
Most esophagi that are short based on preoperative imaging are actually of normal length. It is therefore helpful to think of the short esophagus, as evaluated in the operating room, as falling into three categories: a true, non-reducible short esophagus; a true but reducible short esophagus; and an apparent short esophagus. Perioperative endoscopic or radiologic studies document that all three groups have a GEJ [gastroesophageal junction] located at or above the hiatus. Both the true reducible and nonreducible short esophagi have sustained enough chronic damage to the esophagus to lead to actual intrinsic shortening, whereas patients with an apparent short esophagus have a normal-length esophagus that is merely accordioned into the distal mediastinum. The only way to differentiate between these types is surgical mobilization of the mediastinal esophagus. In most patients (i.e., true, reducible short esophagus and apparent short esophagus), it is possible to reduce the GEJ to at least 2.5 cm below the hiatus. However, in a few patients (i.e., true, nonreducible short esophagus), intraabdominal reduction cannot be accomplished despite extensive transmediastinal or transthoracic esophageal mobilization.
Posterolateral (Bochdalek) Defects
The posterolateral (Bochdalek) defect begins at the vertebrocostal trigone, above and lateral to the left lateral arcuate ligament (Fig. 5). At the time of intestinal return to the abdomen, this trigone is membranous, with few muscle fibers; even at maturity it is variable in size and degree of muscular development. Spreading of the muscle fibers permits a defect, the foramen of Bochdalek, to form and spread anteriorly on the dome of the diaphragm to include the site of the embryonic pleuroperitoneal canal.
The defect can be as small as 1 cm in diameter, or it might involve almost the entire hemidiaphragm. It is much more common on the left side. During an operation, usually no hernial sac is found. The small intestine, stomach, colon, or spleen can be present in the thorax at birth. The lung on the affected side is usually hypoplastic (Fig. 6).
Bilateral congenital posterolateral diaphragmatic hernia is extremely rare and difficult to diagnose.
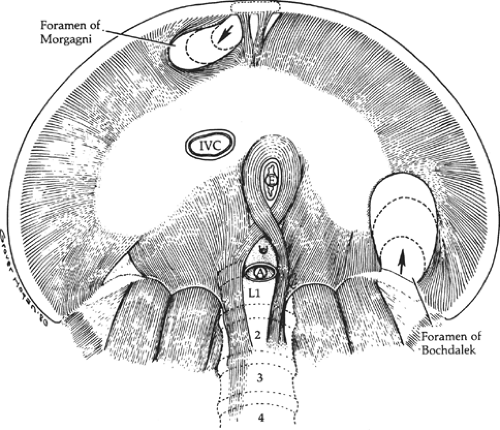 Fig. 5. The diaphragm from below showing the foramen of Bochdalek and the foramen of Morgagni. Both are weak areas of potential herniation. Arrows indicate the direction of enlargement after herniation has begun. A, aorta; E, esophagus; IVC, inferior vena cava |
Parasternal (Morgagni) Defects
Between attachments of the diaphragm to the xiphoid process and the seventh costal cartilage, there is a small gap in the musculature on either side of the xiphoid process (foramina of Morgagni). Herniation at these sites represents only approximately 3% of surgically treated hernias of the diaphragm. The gaps are filled with fat, and the superior epigastric arteries and veins pass through them (Fig. 7).
Herniation through these muscular gaps is almost always the result of postnatal trauma. The hernia occurs more often on the right side, and the herniated organs are usually the omentum, colon, and, eventually, stomach. A sac can be present, or it might have ruptured and disappeared. There might be a predisposition to herniation in those who have large foramina or more fat between muscle fibers, but this pattern has not been demonstrated. Omental herniation through the foramen of Morgagni has been reported. Laparoscopic procedures have been used to repair this defect.
Eventration of the Diaphragm
Congenital eventration describes the abnormal elevation of one leaf of the diaphragm. The entire leaf bulges upward, in
contrast to the localized defect of a foramen of Bochdalek hernia (Fig. 8). The left side is affected more often than the right, and males are affected more often than females. The phrenic nerve appears normal, but the eventrated leaf can consist of a fascial layer with few or no muscle fibers between the pleura and peritoneum. The failure is of muscularization rather than fusion of embryonic components. Intestinal malrotation is often associated. The lung is usually partially collapsed but not hypoplastic; the mediastinum is shifted to the contralateral side, which further reduces ventilation.
contrast to the localized defect of a foramen of Bochdalek hernia (Fig. 8). The left side is affected more often than the right, and males are affected more often than females. The phrenic nerve appears normal, but the eventrated leaf can consist of a fascial layer with few or no muscle fibers between the pleura and peritoneum. The failure is of muscularization rather than fusion of embryonic components. Intestinal malrotation is often associated. The lung is usually partially collapsed but not hypoplastic; the mediastinum is shifted to the contralateral side, which further reduces ventilation.
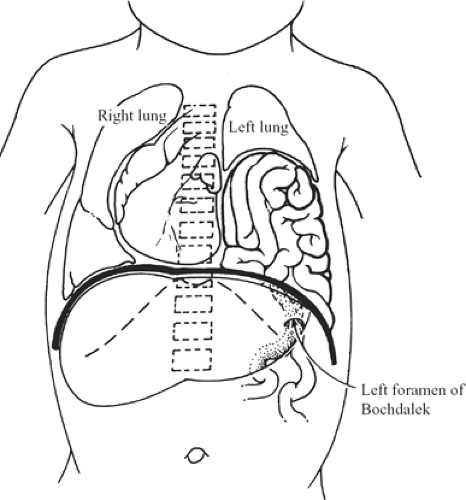 Fig. 6. Herniation of intestines through the foramen of Bochdalek compressing the left lung. The mediastinum is shifted to the right, also reducing the volume of the right lung. |
By contrast, acquired eventration is the result of phrenic nerve injury with normal musculature. The acquired lesion can be temporary; the congenital lesion is permanent unless repaired.
Eventration can be unilateral or bilateral and can rupture in later life. Eventration and acute gastric volvulus have been seen in pediatric patients.
A common but little-understood lesion that has been reported in stillborn infants is ascent of the stomach into the thorax through the esophageal hiatus of the diaphragm. Its congenital origin is not well established.
Peritoneopericardial Hernia
Peritoneopericardial hernia is a rare hernia that lacks a simple embryologic explanation. It has been reported in newborn infants and in adults. A hernial sac, or the trace of one, has been found in a few instances. Because it is in the central tendon and the overlying pericardium, the defect originates in that part of the diaphragm that is formed by the transverse septum.
Liver herniation into the pericardium through the central tendon has been reported. Investigators have found that symptoms of diaphragmatic hernia might not appear until viscera incarcerate in it years after a causal injury.
Other Anomalies Associated with Diaphragmatic Defects
Diaphragmatic anomalies can be associated with other congenital anomalies, such as Cantrell’s pentalogy, tracheal agenesis, genetic syndromes with omphalocele, gastroschisis, intestinal atresias and stenoses, and obstructive uropathies.
Roentgenographic findings support the hypothesis that the cardiac mass is responsible for the caudad displacement of the related hemidiaphragm; this finding is contrary to the classic teaching that it is the liver that lifts the corresponding hemidiaphragm.
The function of the diaphragm, which comprises skeletal voluntary muscle, is as automatic as the function of the heart, with the difference that the cardiac myocytes function as a syncytium. It has been suggested that the diaphragm is second in importance only to the heart in maintaining life.
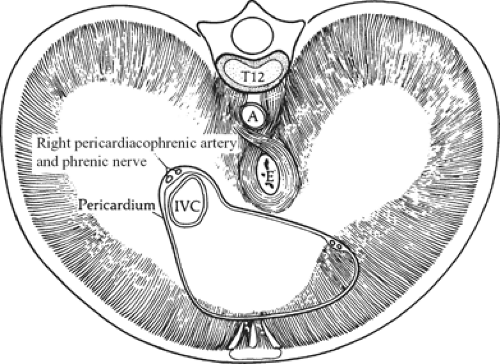 Fig. 7. The diaphragm viewed from above. The area in contact with the pericardium is indicated. The pericardial fibrous tissue is continuous with that of the diaphragm. A, aorta; E, esophagus; IVC, inferior vena cava. |
Pediatric Diaphragm
The following observations have been made from anatomic and ultrasonographic studies of the diaphragm in newborn infants:
The diaphragm inserts only on the anterior costodiaphragmatic rib cage border.
From anterolaterally to posteriorly, the diaphragmatic insertion has increasingly greater distance from the rib cage.
The dorsal diaphragm ends its free course at the 11th rib and continues caudally,
ending between the 12th rib and the crista iliaca. The diaphragm in the newborn acts as a bellows moving mainly in the posterior part, whereas in the adult it acts as a piston (Fig. 9). The diaphragm of the newborn, which has a flat curve because of its large angle of insertion on the rib cage and small area of apposition, has only one physiologic destiny—to suck in the rib cage rather than air. It is this rib cage action that reduces the area of apposition and results in an increase in chest volume.
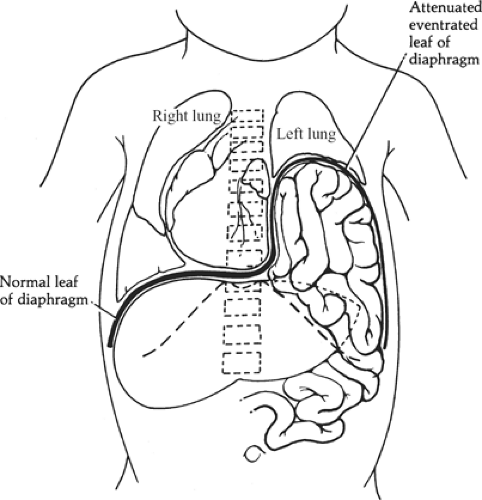 Fig. 8. Eventration of the left diaphragm. The herniated abdominal organs remain beneath the attenuated but intact leaf of the diaphragm. Both lungs are compressed, and mediastinum is shifted to the right. Compare with Figure 6. |
Origins and Insertions of the Diaphragmatic Musculature
The diaphragm comprises a central tendinous area from which muscle fibers radiate in all directions toward their peripheral attachments (Fig. 10).
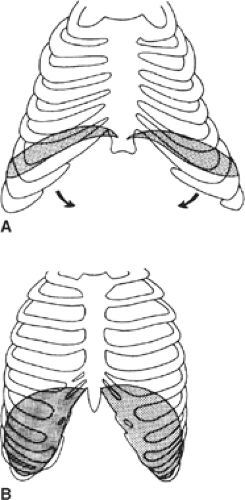 Fig. 9. Developmental changes of the rib cage and the anterior and lateral diaphragmatic insertions from birth (A) to adulthood (B). The stippled surface represents the anterior projection of the diaphragm. (From Devlieger H, Daniels H, Marchal G, et al. The diaphragm of the newborn infant: anatomical and ultrasonographic studies. J Dev Physiol 1991;16:511, with permission.) |
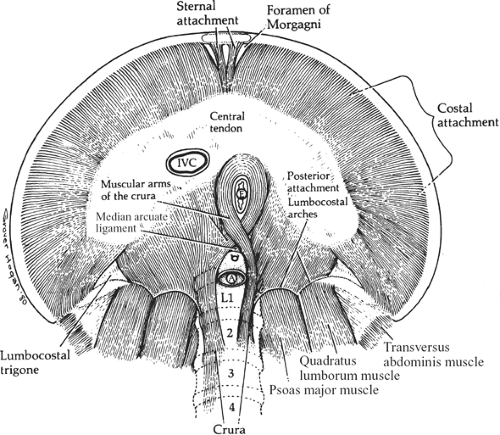 Fig. 10. The attachment of the muscles of the diaphragm seen from below. A, aorta; E, esophagus; IVC, inferior vena cava. |
Sternal Portion
Paired slips of muscle originate from the xiphoid process and the aponeurosis of the transversus abdominis muscle. Small triangular spaces (foramina of Morgagni) separate those slips from the costal fibers and from each other (Figs. 5 and 10). These small triangular gaps are filled with connective
tissue and transmit the superior epigastric vessels (end branches of the internal thoracic (mammary) arteries) and lymphatics that run along them. The sternal portion of the diaphragm arises from the xiphoid process and from the aponeurosis of the transversus abdominis.
tissue and transmit the superior epigastric vessels (end branches of the internal thoracic (mammary) arteries) and lymphatics that run along them. The sternal portion of the diaphragm arises from the xiphoid process and from the aponeurosis of the transversus abdominis.
Costal Portion
Muscle fibers arise from the cartilages of the 7th and 8th ribs, the cartilage and bony portions of the 9th rib, and the distal bony portions of the 10th to 12th ribs.
Lumbar Portion
Posteriorly, the diaphragmatic muscle arises from the crura and the medial and lateral arcuate ligaments (lumbocostal arches). Between the costal and lumbar portion of t he diaphragm is the lumbocostal trigone (Fig. 10), filled in only with connective tissue.
Surgical Considerations
Hollinshead has pointed out that the sternocostal part of the diaphragm is in “intimate” relation to the transversus abdominal muscle. They blend and attach together to the lower six costal cartilages. Therefore, they can be separated together from the ribs. The transversal muscle, which is then made continuous with the diaphragm, can be utilized to help in closure of an anterior defect of the diaphragm.
The lumbar part of the diaphragm is in conjunction with the aponeurosis of the transversus muscle, which blends with the deep layer of the thoracodorsal (lumbosacral) fascia. The latter fuses with the anterior layer medially. Therefore, the latissimus dorsi can be of use in closure of posterior defects of the diaphragm.
Crura
The crura arise from the anterior surface of L1 to L4 on the right, and L1 to L2 or L3 on the left, as well as from the intervertebral disks and the anterior longitudinal ligament. The crural fibers pass superiorly and anteriorly, forming the muscular arms that surround the openings for the aorta and the esophagus; they insert on the central tendon. The crura are tendinous at their origin on the vertebrae, becoming increasingly muscular as they ascend into the diaphragm proper (Fig. 11). In our studies of cadavers, we found the crura to be tendinous, posteriorly and medially, from their vertebral origins to the level of the 10th thoracic vertebra in 90% of cadavers. Sutures to approximate the crura should always be placed through the tendinous portions.
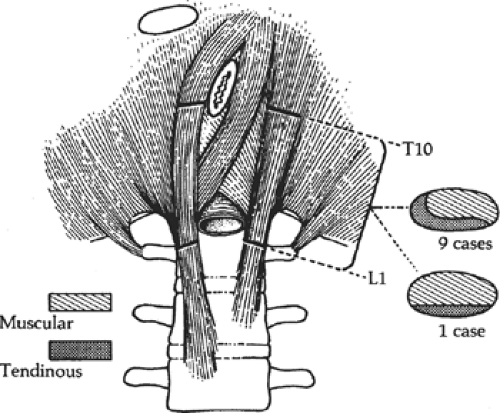 Fig. 11. The crura consists of both tendinous and muscular tissue; only the tendinous portion holds sutures. In 9 of 10 cases, the medial edge of the crura is tendinous. (From Gray SW, Rowe JS Jr, Skandalakis JE. Surgical anatomy of the gastroesophageal junction. Am Surg 1979;45:575, with permission.)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|