Chapter 8 Superficial and deep perivascular inflammatory dermatoses
Chronic superficial dermatitis
Clinical features
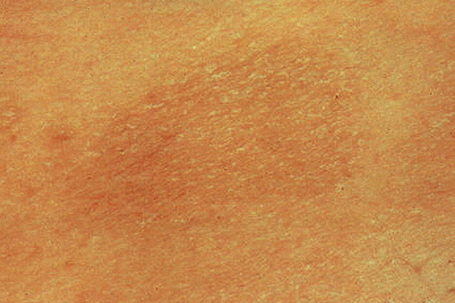
Chronic superficial dermatitis (digitate dermatosis, superficial scaly dermatitis, small-plaque parapsoriasis, persistent superficial dermatitis) is a not uncommon condition, which presents as erythematous scaly persistent patches, showing a predilection for the limbs and trunk. While the lesions may be round or oval, they often have a finger-like appearance, hence the alternative designation of digitate dermatosis (Figs 8.1, 8.2).1 The patches are usually a few centimeters in greatest dimension, but may sometimes be much larger. They are associated with a fine ‘cigarette-paper’ scale that often has a pale white, tan or yellowish color (Fig. 8.3). The disorder is most commonly encountered in middle-aged adults and shows a predilection for men. The patient is usually otherwise asymptomatic. Coexistence with ulcerative colitis has been reported in one patient.2 Lesions tend to chronicity, often persisting for many years. While development of mycosis fungoides has been reported in a patient with chronic superficial dermatitis, it is most likely that these represent separate disease processes rather than disease progression.3
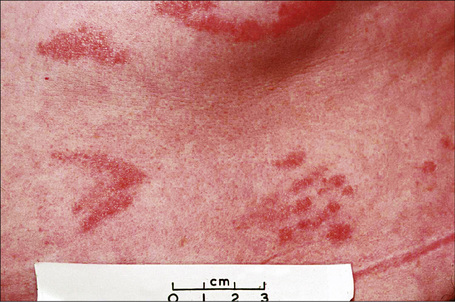
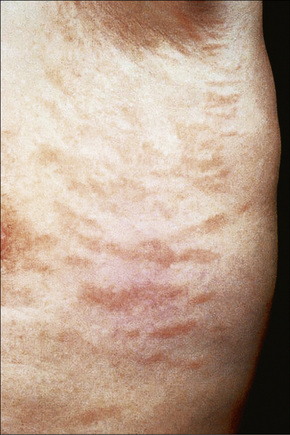
Fig. 8.2 Chronic superficial dermatitis: these uniform, linear lesions had been present for many years.
By courtesy of the Institute of Dermatology, London, UK.
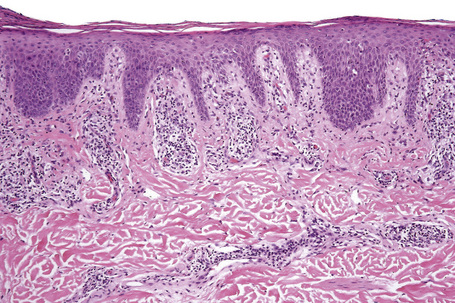
Histological features
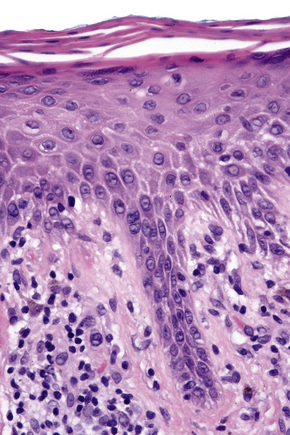
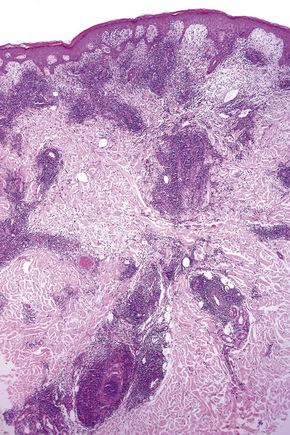
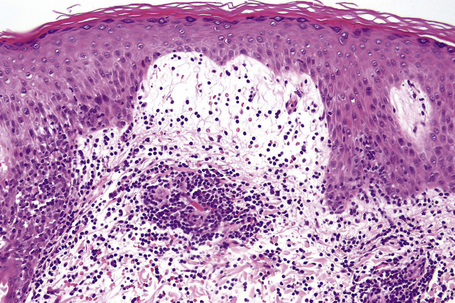
Biopsy shows a superficial perivascular lymphocytic infiltrate (Figs 8.4, 8.5). The infiltrate is of variable density but is often very sparse. Cytological atypia is absent. The epidermis often shows foci of spongiosis. A confluent linear band of parakeratosis spanning multiple rete ridges is a characteristic finding.
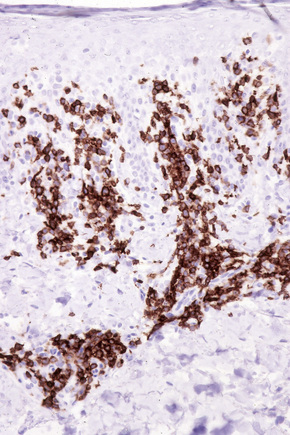
The infiltrate is largely composed of CD4+ T lymphocytes with a minor population of CD8+ T-suppressor cells (Fig. 8.6).2 In a small series, the CD4 to CD8 ratio ranged from 2 to 4.4. The T cells are generally reactive for CD2, CD3, and CD5 (Fig. 8.7). CD7 expression is variable and may be absent. Scattered CD68 reactive macrophages and CD1+ Langerhans cells can be seen.
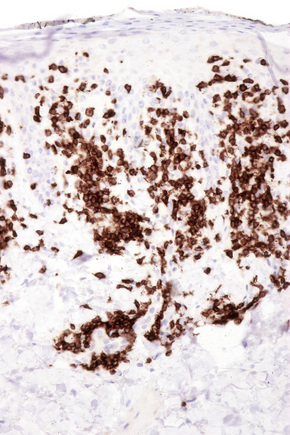
Fig. 8.6 Chronic superficial dermatitis: the infiltrate is composed predominantly of CD4+ T-helper cells.
Differential diagnosis
Immunohistochemistry should be viewed with caution. Loss of T-cell expression may support a diagnosis of mycosis fungoides provided there are histological features in favor of the diagnosis and if the clinical context is appropriate. Loss of CD7 expression, however, may be seen in reactive conditions and this feature is therefore not reliable. Occasionally, only careful review of the clinical information, taken in conjunction with the histological features of previous biopsies (if available) allows for definitive diagnosis. It is important to note that some investigators have demonstrated cases of chronic superficial dermatitis with clonal T-cell gene rearrangements by polymerase chain reaction (PCR).4 One case with a clonal T-cell population resolved, underscoring the growing appreciation that clonality and malignancy are not necessarily synonymous.4 Therefore, it appears that demonstration of a clonal T-cell population may not suffice to reliably distinguish chronic superficial dermatosis from early mycosis fungoides in all cases. In the past, others have concluded that chronic superficial dermatitis is mycosis fungoides.5 The observation that chronic superficial dermatitis rarely, if ever, evolves into (or declares itself as) frank mycosis fungoides has led some authors to cast doubt on this view.6 More recent publications have asserted that chronic persistent dermatitis does not progress to mycosis fungoides.7 It is perhaps more likely that some cases of very early mycosis fungoides cannot be reliably distinguished from chronic superficial dermatitis. Recently, clonal T-cell gene rearrangements have been demonstrated in circulating lymphocytes in blood but not in skin of patients with digitate dermatitis.8 Clearly, long-term follow-up studies are necessary to resolve the significance of clonality in putative cases of chronic superficial dermatitis.
Pityriasis lichenoides may also be confused with chronic superficial dermatitis. Spongiosis without interface changes favors the latter. Pityriasis lichenoides is associated with either vacuolar or lichenoid interface changes in the absence of spongiosis.9
Toxic erythema
Toxic erythema, annular erythema, and gyrate erythema are terms used by dermatologists to describe a number of diseases that share common clinical and histological appearances. Clinically, the terms imply annular erythematous lesions. Pathologists often also use these same terms (particularly gyrate erythema) in a generic manner to describe an inflammatory lesion with a ‘cuffed’ perivascular lymphocytic infiltrate. Although such nomenclature may be used as a descriptor (as one might use terms such as ‘lichenoid’, for example), it should not be taken to imply a specific disease. It is likely that the earlier literature frequently classified different diseases together under these appellations.1 Therefore, to avoid confusion, it is encouraged that these terms are not used in referring to specific diseases.
Erythema annulare centrifugum
Clinical features
Many of these associations are likely to be coincidental and, in most cases, no underlying etiology is identified.31–35 It is unclear whether erythema annulare centrifugum is a distinctive entity or simply represents the clinical expression of a number of inflammatory dermatoses such as hypersensitivity reactions sharing common histological features.36
Mahood and colleagues emphasized that many earlier reports of neoplasia associated with erythema annulare centrifugum are questionable since different subtypes of annular erythemas were often classified together.31,37,38 However, more recent cases have documented erythema annulare centrifugum in patients with underlying malignancy, once again raising the issue of an association with neoplasia. Erythema annulare centrifugum in patients with non-small cell lung carcinoma, carcinoma of breast, colon and prostate, chronic lymphocytic leukemia, and Hodgkin’s lymphoma have been reported in the last decade.14,39–44 In a recent large series, carcinoma was present in 6 of 66 (13%) patients.45 Of these, two had leukemia (acute myelogenous and acute lymphoblastic); one patient had non-Hodgkin’s lymphoma; and three cases were associated with carcinoma (lung, rectal, and hepatocellular).45
Erythema annulare centrifugum has been reported in all age groups, including infants and neonates, but is most commonly seen in young adults.46,47 A recent large series found that the lower extremities, particularly the thighs, were the most frequent site of involvement.45 Nearly 50% of patients in this series had lower extremity involvement. The trunk was affected in 28% of patients and the upper extremity in 16%. The hands, feet, and face are usually spared. Head and neck involvement was seen in only 8% of patients. Laboratory investigation sometimes reveals a peripheral eosinophilia.31 Although individual lesions persist for weeks to a few months before resolving, a course of relapses and remissions over months to years is common and an annually and seasonally recurring form has recently been reported.48 Kim et al. found that lesions lasted from 3 days to 18 years with a mean duration of 2.8 years.45
The lesions take the form of annular erythematous bands, which may spread outwards or remain stationary (Figs. 8.8, 8.9). They are well circumscribed with raised edges, and slight scaling that tends to trail behind the advancing margin.49 With time, central clearing is seen. Arcuate and polycyclic variants are therefore occasionally evident.31 Lesions may be mildly pruritic. Vesiculation is rare.32
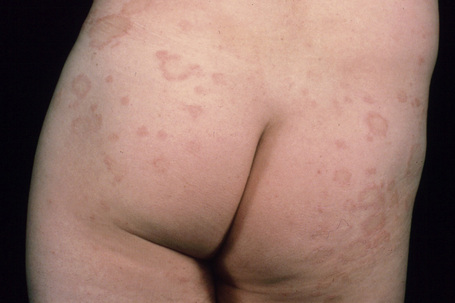
Fig. 8.8 Erythema annulare centrifugum: bilateral annular lesions are present on the buttocks.
From the collection of the late N.P. Smith, MD, The Institute of Dermatology, London, UK.
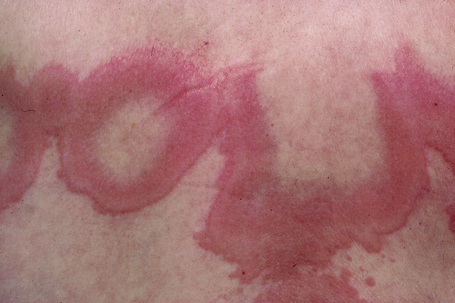
Fig. 8.9 Erythema annulare centrifugum: close-up view.
From the collection of the late N.P. Smith, MD, The Institute of Dermatology, London, UK.
Some authors have divided the disease into two distinct subtypes: superficial and deep gyrate erythema.45
Histological features
A spectrum of non-specific histological findings is seen in erythema annulare centrifugum. As noted above, deep and superficial variants are recognized.45
In the superficial variant, a well-demarcated perivascular infiltrate of lymphocytes and histiocytes, often described as having a ‘coat sleeve’’ or ‘pipe-stem’ appearance, is confined to the superficial dermis (Figs 8.10, 8.11). The overlying epidermis often may be normal; however, epidermal changes including mild spongiosis, slight and focal basal layer vacuolar degeneration, mounds of parakeratosis or hyperkeratosis are encountered in approximately 50% of patients.14,45
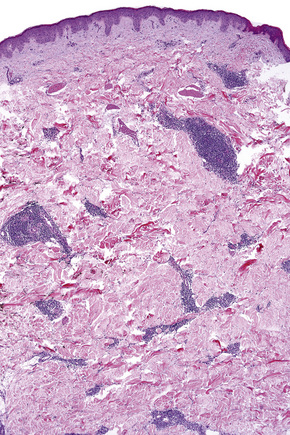
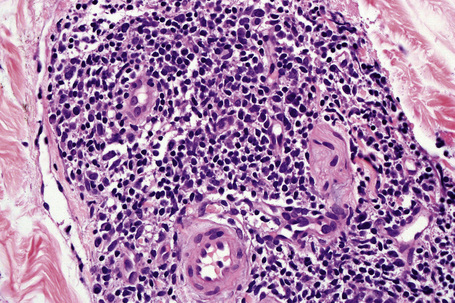
Fig. 8.11 Erythema annulare centrifugum: the infiltrate is composed of mature lymphocytes and histiocytes.
In the deep subtype of erythema annulare centrifugum, the perivascular infiltrate involves both the superficial and deep plexuses.14,32–34,45 Epidermal changes are usually absent or minimal.
Erythema gyratum repens
Clinical features
Erythema gyratum repens (L. repens, to crawl or creep) is an extremely rare and clinically distinctive figurate eruption usually associated with an underlying malignancy. The most commonly associated neoplasm is carcinoma of the lung; other affiliated tumors include carcinoma of the uterus and cervix, esophagus, stomach, kidney, and breast as well as essential thrombocythemia.1–11 Treatment of the cancer may be associated with remission of the cutaneous eruption, while tumor recurrence or metastases can be accompanied by a relapse.2 Rarely, the condition develops in the absence of an underlying malignancy.12–17 It may disclose underlying pulmonary tuberculosis. In one patient with no evidence of malignancy, the rash resolved a few days after removal of a cavitary tuberculoid lung lesion.14
Ichthyosis may accompany erythema gyratum repens.18 A report of a patient with transitional cell carcinoma of the kidney who developed erythema gyratum repens and ichthyosis comes as no surprise since both conditions are associated with malignancy. The combination of ichthyosis, palmoplantar keratosis, and erythema gyratum repens, in the absence of malignancy, has also been reported.19
Erythema gyratum repens-like eruptions have also been described in the presence of connective tissue diseases. Typical erythema gyratum repens developed in a patient with cutaneous subacute lupus erythematosus following hydroxychloroquine treatment.20,21 The authors of this report concluded that the patient’s rash represented a peculiar pattern of involvement by subacute lupus which they designated subacute lupus gyratum repens. An erythema gyratum repens-like eruption has also been described in association with Sjögren’s syndrome, neutrophilic dermatosis, leukocytoclastic vasculitis, in patients with lupus erythematosus, and in the setting of urticarial vasculitis.22–25
Caputo et al. reported linear IgA dermatosis, erythema, and an eruption resembling erythema gyratum repens in a patient without malignancy.26 Bullous pemphigoid may be associated with erythema gyratum repens and an erythema gyratum repens-like eruption has been documented in a patient with treated and resolving psoriasis and with epidermolysis bullosa acquisita accompanied by ulcerative colitis.27–31
Erythema gyratum repens has been described in patients with hypereosinophilic syndrome, also with no evidence of neoplasia.32
The eruption, which may precede the malignancy by months, takes the form of concentric bands of erythema in an annular or gyrate arrangement (Figs 8.12, 8.13). These bands have been described as having a ‘timber grain’ or ‘zebra-like’ pattern and they move (up to about 1 cm) daily.33 Scaling occurs and there may be pruritus. Lesions often commence on the arms and legs, but frequently become generalized.1 The hands, feet, and face are usually spared.33 Postinflammatory hyperpigmentation may be a feature.1 Hyperkeratosis of the palms and soles is also sometimes present.3,12 Males are affected twice as commonly as females.3 Patients are usually in their seventh decade.20
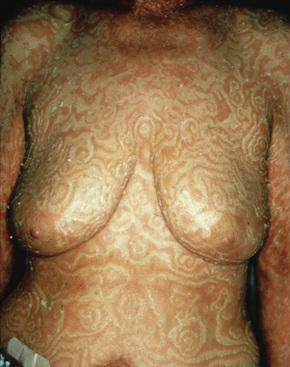
Pathogenesis and histological features
Erythema gyratum repens may have an immunological pathogenesis, since granular deposits of IgG and C3 have been found at the basement membrane zone of both involved and uninvolved skin in a patient with associated bronchial carcinoma and in involved non-sun-exposed skin in another unassociated with neoplasia.16,34–36 In a separate patient, although basement membrane zone immunofluorescence was negative, epidermal nuclear labeling was identified.37 Caux et al. reported one patient with squamous cell carcinoma of the lung who had immunoreactants at the basement membrane of involved and normal non-sun-exposed skin. In addition, this patient showed staining of IgG, IgM, and C3 along the basement membrane of the bronchus.35 However, the immunoreactants did not localize to the tumor.
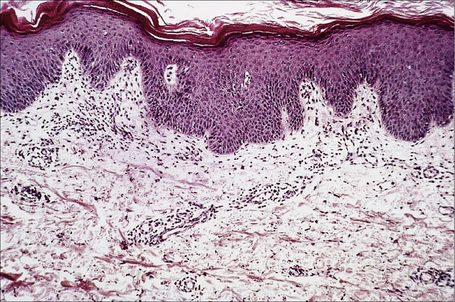
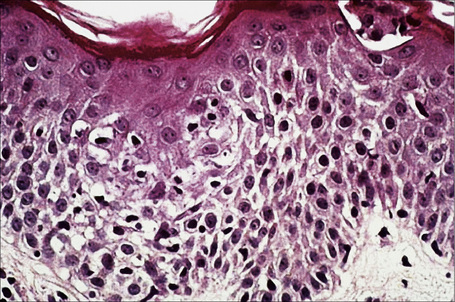
The appearances in erythema gyratum repens are not diagnostic. They include hyperkeratosis, parakeratosis, acanthosis, and spongiosis, together with a superficial perivascular lymphohistiocytic infiltrate in the papillary dermis (Figs 8.14, 8.15).2
Lymphocytic infiltrate of the skin (Jessner)
Clinical features
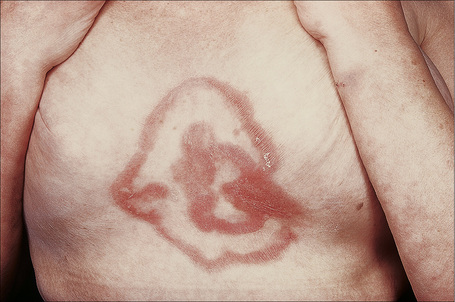
Jessner’s lymphocytic infiltrate of the skin is an uncommon dermatosis of unknown etiology, although a relationship with sun exposure, at least in the early stages, is occasionally documented.1 Lesions, which may be single or more often multiple, occur most often on the face, neck, back, and upper chest, and present as 1–2-cm diameter, asymptomatic, discoid or annular, erythematous or brownish papules or plaques that often show central clearing to produce circinate lesions (Figs 8.16, 8.17).1–4 Familial cases have occasionally been documented.5–8
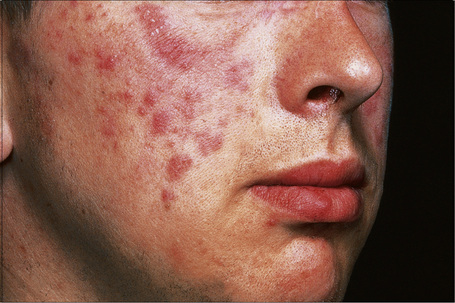
Fig. 8.16 Jessner’s lymphocytic infiltrate: there are multiple erythematous plaques on this young man’s cheek.
By courtesy of the Institute of Dermatology, London, UK.
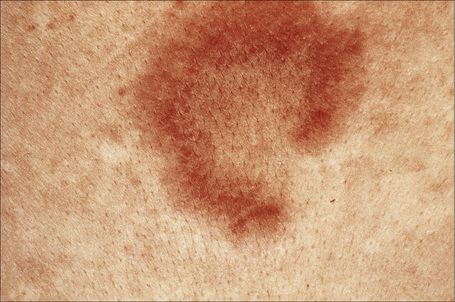
Fig. 8.17 Jessner’s lymphocytic infiltrate: central clearing has resulted in this circinate lesion.
By courtesy of the Institute of Dermatology, London, UK.
In contrast to discoid lupus erythematosus, with which it is sometimes confused, there is no hyperkeratosis, telangiectases or follicular plugging, and scarring is not a feature. Rarely, however, the two diseases appear to coexist.1 The disease tends to affect adults, particularly in the third to fifth decades. Although some authors have found a predilection for males, 54% of patients in a large series were female and the overall gender distribution appears to be equal.1,3 Rarely, the condition presents in children.9–11 Lesions often resolve within weeks or months, but relapses are not uncommon and, in many patients, the disorder persists for years. The eruption is not characterized by seasonal variation. The evidence available suggests that lymphocytic infiltrate of the skin is a distinctive dermatosis. It does not evolve into lupus erythematosus, polymorphous light eruption or lymphoma.1
Pathogenesis and histological features
Braddock and coauthors found that natural killer cell lytic activity and antibody-dependent cell-mediated cytotoxicity was decreased.10 This same group identified increased levels of circulating immune complexes in patients with lymphocytic infiltrate of skin. In two patients immune complexes decreased to normal levels following treatment but became elevated during recurrence of disease following treatment.10,11 Based on these observations, these investigators concluded that immune defects might be important in the pathogenesis of Jessner’s lymphocytic infiltrate. Of interest, similar findings have been observed in patients with reticular erythematous mucinosis. Clearly, further study is necessary to determine the pathogenesis of this disease.
The epidermis is typically unaffected. Within the superficial and mid dermis is a perivascular and, much less commonly, a perifollicular infiltrate of mature lymphocytes (Figs 8.18, 8.19). Occasional histiocytes and scattered plasma cells may also be present and sometimes there is an increase in dermal ground substance.12 Lymphoid follicles are not a feature.
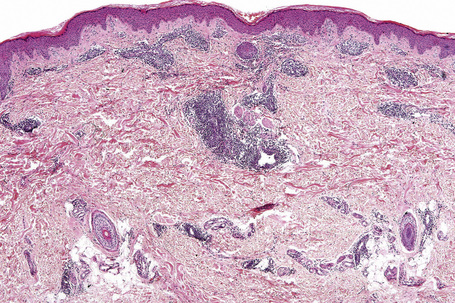
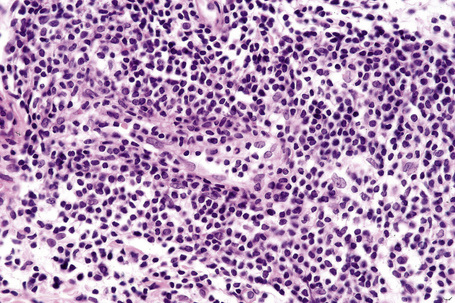
Fig. 8.19 Lymphocytic infiltrate of Jessner: the infiltrate is composed almost entirely of small lymphocytes.
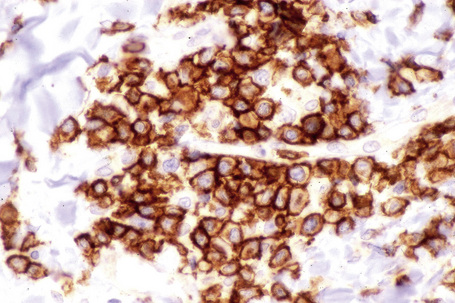
The infiltrate consists predominantly of T cells, most often of the CD4+ helper subtype (Fig. 8.20). Occasionally, however, CD8+ suppressor T cells constitute the majority of cells.3,13–16 Leu 8 is commonly expressed but human leukocyte antigen (HLA)-DR is not present. B cells are relatively sparse in number or are absent.
Differential diagnosis
Lymphocytic infiltrate of the skin differs from discoid lupus erythematosus by the absence of epidermal changes, scarring, and a negative lupus band test. Immunohistochemistry may sometimes be helpful. The infiltrate in lymphocytic infiltrate is HLA-DR negative in contrast to discoid lupus erythematosus in which the lymphocytes and often the keratinocytes are HLA-DR positive.17 Leu 8 (immunoregulatory T-cell) expression is also more frequently seen in lymphocytic infiltrate.13,18 In one study, the average percentage of Leu 8 positive lymphocytes was 65% in lymphocytic infiltrate of skin and only 15% in discoid lupus erythematosus.18 The presence of CD20+ B cells favors lupus erythematosus, which tends to be composed of a mixture of B and T cells. In contrast, T cells predominate in lymphocytic infiltrate of skin.14,19,20 One group of investigators has suggested that the presence of plasmacytoid monocytes favors a diagnosis of lymphocytic infiltrate of skin over lupus erythematosus. They found plasmacytoid monocytes to be present in 58% of patients with lymphocytic infiltrate of skin but only in 7% of patients with discoid lupus erythematosus.21 Others, however, have not been able to corroborate this finding.19 The presence of significant dermal mucin would support lupus erythematosus. Reliable distinction between lymphocytic infiltrate of the skin and tumid lupus erythematosus may be difficult and frequently impossible since lymphocytic infiltrate may sometimes be accompanied by excess dermal mucin. It has therefore been speculated that these entities represent a continuous disease spectrum rather than different diseases.22 The finding that a subset of patients with lymphocytic infiltrate of the skin also had a confirmed diagnosis of lupus erythematosus lead one group to speculate whether lymphocytic infiltrate of the skin could represent a variant of lupus erythematosus.4
Epidermal Langerhans cells are often increased in lymphocytic infiltrate whereas they are frequently reduced in number in discoid lupus erythematosus.13
Lymphocytic infiltrate is usually histologically indistinguishable from polymorphous light eruption although early lesions of the latter may show edema of the papillary dermis. It should be noted, however, that sometimes the two conditions may coexist. In cases where the diagnosis is in doubt, phototesting may be necessary. One group of investigators has found the presence of plasmacytoid monocytes to favor lymphocytic infiltrate of the skin over polymorphous light eruption.21 However, this has not been confirmed (see above).
Reticular erythematous mucinosis
Clinical features
This rare chronic dermatosis, which shows a female predominance (2:1), has been described worldwide.1 Although it may affect a wide age range, it most frequently develops in the second to fourth decades.2 Rarely, it is encountered in children.2 Familial presentation is exceptional.3 It usually presents as a persistent, reticulate, urticated, macular, and sometimes papular, erythema with an irregular, but well-defined border. The lesions typically occur on the central chest and upper back (Figs 8.21–8.23).4–6 Less commonly, they can be found on the face, arms, abdomen, and groins, but the peripheries are spared.1,7,8 Patients frequently notice an exacerbation in the sun, but the relationship between sunlight and the disease (if any) is not well understood.2,6–11 Although patients are usually asymptomatic, some report pruritus or burning following exposure to sunlight. There is no evidence of systemic involvement.
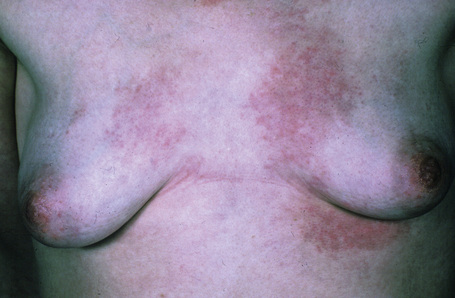
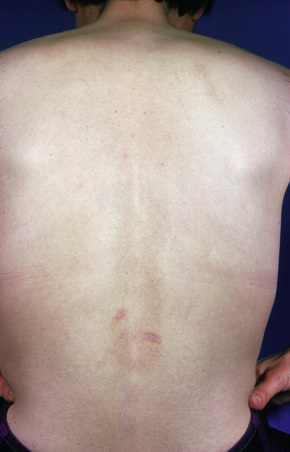
Fig 8.22 Reticular erythematous mucinosis: location in the lower back is unusual.
From the collection of the late N.P. Smith, MD, The Institute of Dermatology, London, UK.
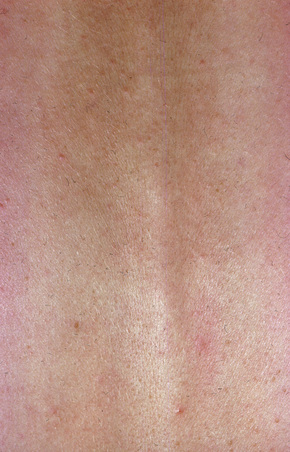
Fig. 8.23 Reticular erythematous mucinosis: closer view of previous figure. Macular elements predominate.
From the collection of the late N.P. Smith, MD, The Institute of Dermatology, London, UK.
Occasional patients have more infiltrated papules and plaques; this was originally described as plaquelike cutaneous mucinosis, but is now accepted as a variant of reticular erythematous mucinosis.12,13 Of particular interest, the plaquelike form of the disease has been documented in association with carcinoma of the breast and colon and one patient suffered from essential thrombocytosis in addition to carcinoma of the lung.13,14
Patients with this condition have an increased risk of thyroid disease, arthritis, and diabetes mellitus.2 In a series of nine patients, one patient had evidence of Hashimoto’s disease while another had hyperthyroidism.13 A patient with reticular erythematous mucinosis and myxedema in the setting of Hashimoto’s thyroiditis has also been described.15
Reticular erythematous mucinosis in patients with human immunodeficiency virus (HIV) infection has also been documented.16,17 Of interest, other cutaneous mucinoses that have been described in association with HIV infection include scleredema and lichen myxedematosus.17 Furthermore, deposition of mucin in the bone marrow of patients with acquired immunodeficiency syndrome (AIDS) is a common finding.18 The pathogenetic relationship between these various forms of mucin deposition, if any, has not been defined. It is, of course, tempting to postulate that they are related but data to support such a conclusion are not yet established.
The presence of monoclonal IgG (kappa) paraproteinemia has been reported in one patient.19
Pathogenesis and histological features
The pathogenesis of reticular erythematous mucinosis is not well understood. Phototoxicity likely plays some role in the disease, either directly or indirectly. Braddock and coauthors found that natural killer cell lytic activity and antibody-dependent cell-mediated cytotoxicity were decreased.20 This same group found increased levels of circulating immune complexes. Of interest, two patients were observed to have circulating immune complexes that decreased with treatment only to become elevated during recurrence of disease following treatment.20 Based on these observations, the investigators concluded that immune defects may be of importance in the pathogenesis. Of interest, similar findings have been observed in patients with Jessner’s lymphocytic infiltrate (see above). Clearly, further study is necessary regarding the precise pathogenetic basis of this disease.
The epidermis may be slightly flattened or appear normal. Within the dermis there is moderate vascular dilatation associated with a marked mononuclear perivascular and often perifollicular infiltrate composed mainly of T-helper lymphocytes (Figs 8.24, 8.25).6,13,20–22 Excess mucin (predominantly hyaluronic acid) is usually present in the upper dermis but in more chronic lesions it is sometimes absent (Fig. 8.26). The mucin stains positively with Alcian blue (pH 2.5) and colloidal iron, but is usually not metachromatic with toluidine blue. The collagen fibers are separated, but appear morphologically normal. Fragmentation of elastic fibers is sometimes a feature.7 There is no evidence of fibroblastic proliferation, but by immunohistochemistry increased numbers of factor XIIIa-positive dermal dendrocytes have been identified.23
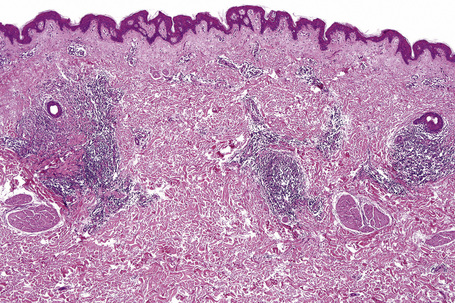
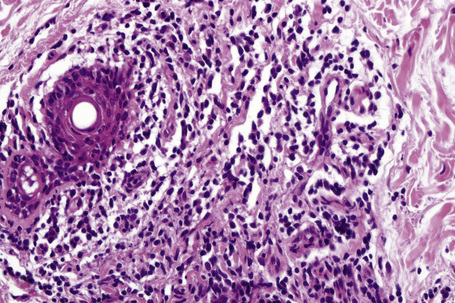
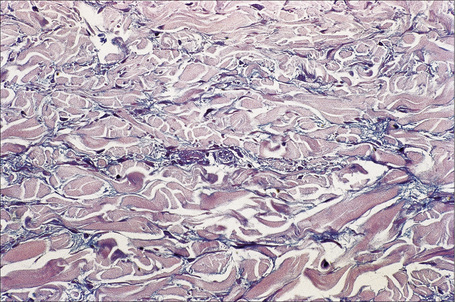
A few cases with positive direct immunofluorescence have been reported. IgM-reactive papillary dermal cytoid bodies were documented in one case.20 Rare examples show staining for IgM along the dermal–epidermal junction.2,10,24 The significance of these findings is unclear but may be further evidence of an immunological basis for the pathogenesis of this condition.24
Ultrastructural studies are largely unhelpful. Other than demonstrating conspicuous and dilated rough endoplasmic reticulum within dermal fibroblasts, electron microscopy merely serves to confirm the light microscopic observation of widely separated fascicles of collagen fibers.21 In a number of reports tubuloreticular structures were identified within the cytoplasm of endothelial cells.7,25,26 Although at one time these were thought to represent paramyxoviruses, more recent studies suggest that they may be derived from infolded endoplasmic reticulum. They have also been identified in pretibial myxedema, lupus erythematosus, dermatomyositis, malignant atrophic papulosis, and various lymphomas.27
Differential diagnosis
The principal clinical and pathological differential diagnoses include lupus erythematosus and polymorphous light eruption. Distinguishing between lupus erythematosus and reticular erythematous mucinosis may be very difficult, particularly as one condition may evolve into the other.28 Histologically, reticular erythematous mucinosis lacks the epidermal changes of lupus erythematosus and the immunofluorescent findings are usually, but not always, negative.7–9 As noted above, there are a few reports in which granular immunoglobulin deposition at the dermoepidermal junction has been identified.2,10,24 The presence of several immunoreactants favors a diagnosis of lupus erythematosus. Clinical and serological studies are also necessary to establish a diagnosis of lupus erythematosus.
In polymorphous light eruption, mucin deposition is much less striking and is limited to the papillary dermis.29 Perifollicular inflammation is not a feature of polymorphous light eruption. In addition, epidermal changes of spongiosis – sometimes with vesiculation in papular and eczematous lesions and mild basal cell hydropic change in the plaque variant – serve as further distinguishing features.30 Polymorphous light eruption resolves once exposure to sunlight has ceased, in contrast to reticular erythematous mucinosis where the lesions persist.
Histologically, reticular erythematous mucinosis also shows some overlap with lymphocytic infiltrate of Jessner.2 Mucin deposition, however, is not generally a feature of the latter condition and the inflammatory cell infiltrate is always more prominent.
Polymorphous light eruption (including juvenile spring eruption and lambing ears)
Clinical features
Polymorphous (polymorphic) light eruption, which is the most common photodermatosis, usually presents in young people as recurrent erythematous papules, vesicles and/or plaques following exposure to ultraviolet (UV) light (Figs 8.27, 8.28).1–6 The face, chest, upper back, and extremities are the most common sites of involvement.5 Most patients have multiple lesions.5 In one study of 138 patients, the mean age at onset was 26 years.5 There is a predilection for young women, with 89% of patients being female.7,8 The vast majority of lesions are associated with pruritus. Most patients require less than 30 minutes of sun exposure to elicit clinical features.5 Onset following light exposure typically takes 18–24 hours. Either the UVA or UVB part of the light spectrum may cause lesions.2,9 Lesions are caused by UVA light in 56% of cases, UVB in 17%, and both UVA and UVB ranges in 26% of cases.2 However, some authors have not been able to elicit lesions with UVB light.4 Exposure resulting in sunburn is not necessary for the development of the condition.4 Some patients report symptoms resulting from light exposure through glass.4
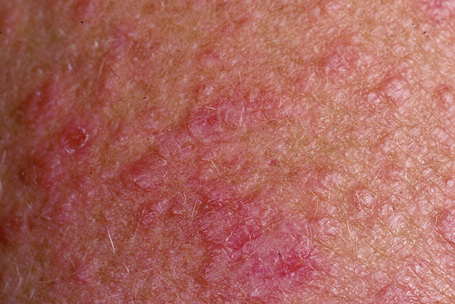
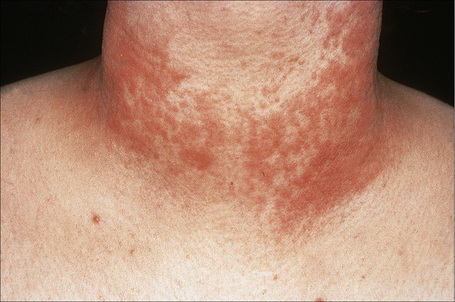
Fig. 8.28 Polymorphous light eruption: the eruption is typically symmetrical and is usually pruritic.
By courtesy of the Institute of Dermatology, London, UK.
Polymorphous light eruption most often occurs in patients with fair skin; however, dark-skinned individuals can also be affected. The disease is more common in people residing in northern latitudes. One study showed that the prevalence rates for London (UK) and Perth (Australia) were 14.8% and 5.2%, respectively.10 In a recent retrospective analysis, however, the incidence of photosensitivity reactions and polymorphous light eruption in particular was found to be roughly comparable for dark-skinned and Caucasian individuals.11 The pinpoint papular variant of polymorphous light eruption has only recently been recognized and appears to be particularly common on dark skin.11,12 It is characterized by numerous grouped, small, 1–2-mm papules, which may be accompanied by small vesicles in the acute phase.13
It appears that the incidence of polymorphous light eruption is much more common than is demonstrated by contact with healthcare workers. In one survey, 21% of workers in a Swedish pharmaceutical company had symptoms consistent with polymorphous light eruption; however, only 3% had sought medical attention for their symptoms.7
Biopsy of experimentally induced lesions shows similar histological features compared to clinical lesions.4 Given the role of sun exposure in its pathogenesis, it comes as no surprise that polymorphous light eruption is most often seen in spring and summer.4 In addition, it is not uncommon for the first sign of disease to manifest during a vacation to southern latitudes. The features, which develop after a latent period of hours to days, commonly subside completely within days and heal without sequelae.1 However, once the disease is established, persistence for many years is common.3 Overall, however, there is diminution of light sensitivity over time, but this process often takes years.3 In a large study, the mean disease duration of the condition was 10.5 years.5 Patients with a duration of up to 53 years have been studied.7 The distribution of lesions often changes with time.5
One study showed that thyroid disease was present in 14% of patients.14 This same study found lupus erythematosus in only 2 of 94 patients. The authors concluded that the risk of lupus erythematosus was not increased in patients with polymorphous light eruption. Authors of another study, however, have suggested that a subgroup of patients with polymorphous light eruption may be at an elevated risk for lupus erythematosus.15
Juvenile spring eruption appears to be either a variant of polymorphous light eruption or a closely related disorder.16–21 In one study, the prevalence was 6.7% with a male predominance.16 The lesions are characterized by erythematous papules and vesicles located on sun-exposed portions of the helix of the ear following light exposure. They tend to be pruritic. In one study, 4 of 18 patients also had lesions of typical polymorphous light eruption.17 As its name implies, the lesions tend to occur in the spring. A positive family history is present in some patients.19 A disorder, clinically and histologically reminiscent of juvenile spring eruption, has also been reported to develop in farmers at the time of lambing and calving. It is provisionally termed ‘lambing ears’.22
Pathogenesis and histological features
The pathogenesis of polymorphous light eruption is poorly understood. Study of adhesion molecule expression has led some authors to propose that polymorphous light eruption is immunologically mediated.23 Specifically, vascular endothelial expression of endothelial leukocyte adhesion molecule-1 (ELAM-1), vascular cell adhesion molecule-1 (VCAM-1), and keratinocyte and endothelial expression of intercellular adhesion molecule-1 (ICAM-1) in biopsies of induced lesions has been documented.23 The authors noted that their results were similar to those seen in delayed-type hypersensitivity reactions. In addition, reduced skin infiltration by neutrophils following UVB exposure was noted.24 However, the triggering antigen(s) are unknown.23 It appears that polymorphous light eruption may be a heritable disorder. In one study, 46% of patients reported a positive family history.7
Histologically, a perivascular lymphohistiocytic infiltrate is present in the superficial and sometimes deep dermis (Figs 8.29, 8.30).25–27 A characteristic, but not uniformly present feature, is papillary dermal edema, which is often marked. The presence of massive papillary dermal edema may be associated with subepidermal or intradermal vesicle formation.
Papular and papulovesicular lesions may show epidermal acanthosis, spongiosis, occasional dyskeratotic cells, and lymphocyte exocytosis.25,27 Spongiosis may sometimes become so severe as to lead to intraepidermal vesicle formation.25 Other authors, however, have not found spongiosis to be a significant feature.26 Basal cell vacuolization, usually mild, is found in some cases.25,27,28 Periadnexal involvement by a chronic inflammatory cell infiltrate may be present in papular and papulovesicular lesions.25 Some authors have reported increased eosinophils and neutrophils; however, others have not confirmed this observation.25,26 Papillary dermal erythrocyte extravasation is commonly present.25 Finally, features secondary to scratching, such as hyperkeratosis and acanthosis, may be seen.27
Immunofluorescence studies have shown that immunoreactants (C3, IgG, and IgM) may be present along the basement membrane zone.3 However, when staining is evident, it is usually weak.3
Juvenile spring eruption of the ears is characterized by a perivascular lymphohistiocytic infiltrate often associated with subepidermal vesicle formation.20
In early lesions, helper-inducer T lymphocytes predominate and increased numbers of dermal Langerhans cells are present.26 With chronicity, cytotoxic suppressor T cells become more conspicuous.
Differential diagnosis
It should be noted that there is considerable variability in both the clinical and histological descriptions of polymorphous light eruption. This has led some authors to suggest that polymorphous light eruption likely represents a group of related disorders rather than a single entity.25,26 Phototesting, therefore, is probably the best ‘gold standard’ for establishing the diagnosis. Compared with reticular erythematous mucinosis, mucin deposition is absent or much less prominent in polymorphous light eruption. Clinically, polymorphous light eruption resolves once exposure to sunlight has ceased in contrast to the persistent lesions of reticular erythematous mucinosis.
Tumid lupus erythematosus
Clinical features
Lupus erythematosus is discussed in detail in Chapter 17 and the reader is referred there for a comprehensive discussion of the disease. In this section only the tumid variant of lupus erythematosus is discussed.
Tumid lupus erythematosus (lupus erythematosus tumidus) is a rare manifestation of lupus that some authors believe to be sufficiently characteristic to justify classification as a distinctive subtype of chronic cutaneous lupus erythematosus.1 However, the lack of an agreed-upon diagnostic gold standard makes this designation somewhat controversial. Further study and refinement of criteria for inclusion into this subtype of lupus and to allow for reliable distinction from other inflammatory dermatoses is necessary.
Raised erythematous plaques, which have been described as ‘succulent’, characterize the clinical lesions.1 Follicular plugging is not a feature.2 Annular and gyrate forms are seen in some patients.1 The sun-exposed areas such as the face, chest, arms, and shoulders are most commonly affected.1–4 In the largest series published to date, patients with this clinical appearance accounted for 16% of the total number of patients seen in a large cutaneous lupus clinic.1 Approximately equal numbers of male and female patients are affected, in contrast to the preponderance of females affected by other subtypes of cutaneous lupus.2,4 In this variant, young adults are most often encountered but presentation in childhood is rare.4,5 In most patients, lesions can be reproduced by exposure to UVA or UVB light.1,3,6 Development of tumid lupus erythematosus has also been reported following highly active antiretroviral therapy in the setting of HIV as a manifestation of immune restoration as well as in the setting of estrogen and infliximab treatment.7–9 Unusual and rare presentations include unilateral distribution in addition to symmetrical involvement of both elbows.10,11
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


