Sterilization in practice
Jean-Yves Maillard and Susannah E. Walsh
Chapter contents
Determination of sterilization protocols
Recommended pharmacopoeial sterilization processes
Steam (under pressure) sterilization
Integrated lethality in sterilization practice
Statistical considerations of sterility testing and sterility assurance level
Test for sterility of the product
Validation of a sterilization process
Limitations of sterilization methods
Key points
Sterile products
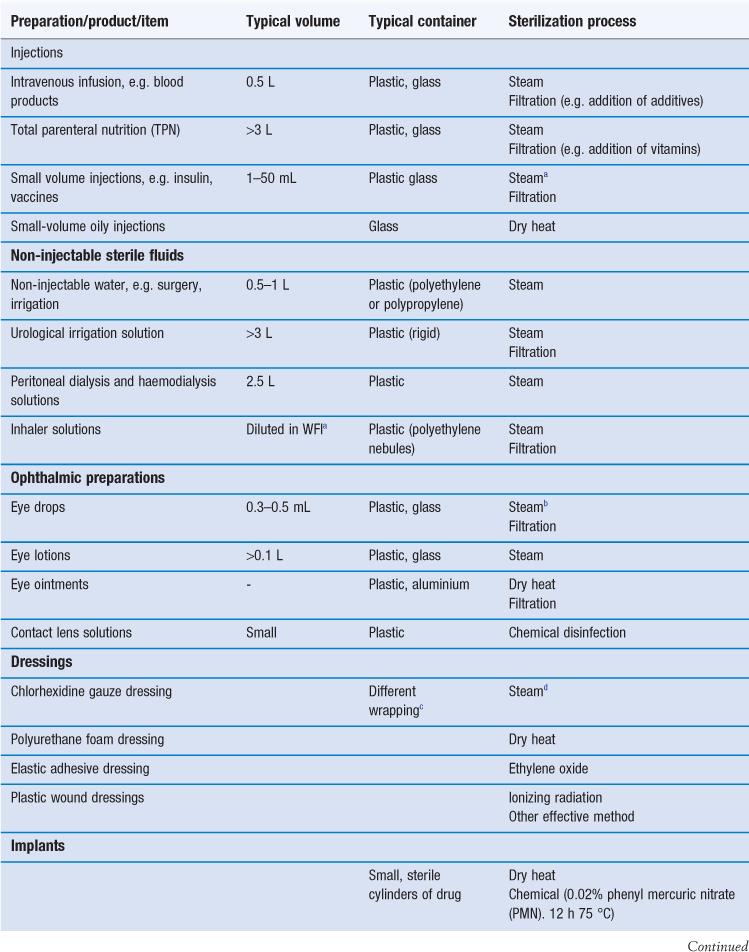
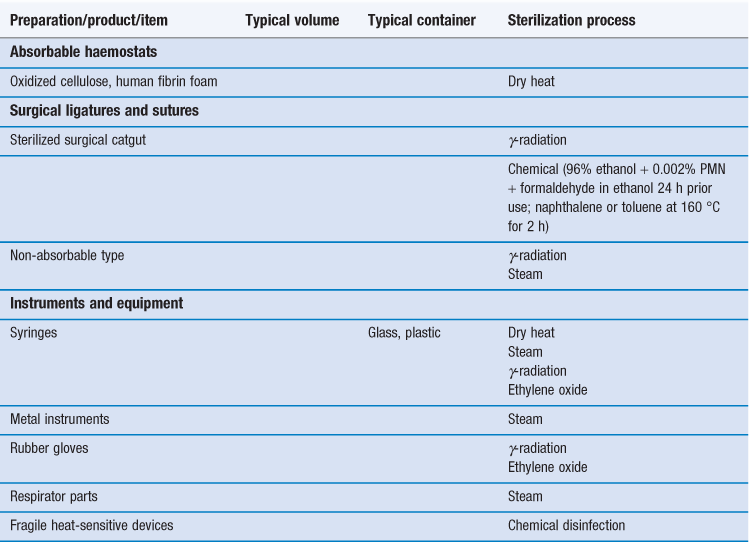
Sterilization is an essential part of the processing of pharmaceutical dosage forms that are required to be sterile. By definition, a sterile product is completely free of viable microorganisms. In addition to the pharmaceutical products that require to be sterile, a number of medical devices that come into contact with sterile parts of the body or are reused in patients also need to be free of microorganisms (Table 17.1). The diversity of the items to be sterilized in terms of properties (e.g. heat sensitive or not), bulk, content, and the number of items to be sterilized per load requires the use of distinct sterilization processes. The British Pharmacopoeia (2013a), as an example, describes the use of five main sterilization processes to accommodate the range of products to be sterilized: steam, dry heat, gaseous, ionizing radiation and filtration sterilization. The first four methods are usually used to process products in their final containers (terminal sterilization). Regardless of the sterilization methodology used, it is important that the process itself is fully validated. A number of guidelines and European/international standard documents for specific product-sterilization method combinations exist and are followed by manufacturers and end users. Failure to control and/or document adequately a sterilization process can lead to serious incidents. This chapter aims to provide a brief overview of the recommended sterilization processes, their control and validation.
Determination of sterilization protocols
There are various technologies available to achieve sterility of pharmaceutical preparations and medical devices (Table 17.2). Generally, sterilization of the product in its final container (terminal sterilization) is preferred. This infers that the container must not impinge on the optimum sterilization to be delivered and that the container and closure maintain the sterility of the product throughout its shelf-life. The selected sterilization process must be suitable for its purpose, i.e. the sterilization of a given product, device and preparation, which means that the product and its container have to be rendered sterile and must not be damaged by the process or post process.

The choice of an appropriate sterilization process depends on a number of factors (Table 17.3) related to the product to be sterilized, such as type and composition of product and also the quantity to be sterilized. Additionally, the composition and the packaging of the product are significant factors that rule out some sterilization processes. For example, a heat-labile preparation would not be sterilized by heat and a small oily injection would not be sterilized by steam sterilization (further examples are given in Table 17.1). For specific types of products such as dressings, although moist heat sterilization is generally the method of choice, only certain types of autoclave, such as vacuum and pressure-pulsing autoclaves are appropriate.
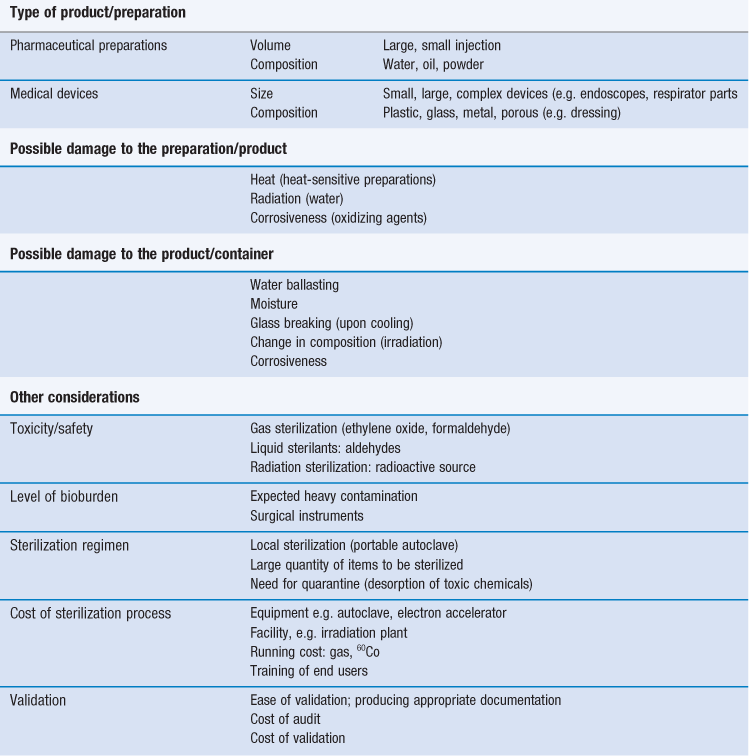
For any given preparation or product, it is difficult to predict the microbial bioburden prior to sterilization. It is assumed that the bioburden of pharmaceutical preparations will be minimal as the manufacturing process should adhere to good manufacturing practice (GMP) (Box 17.1). However, a sterilization process should be able to deal with a worst case scenario. This is usually exemplified by the use of biological indicators (see the ‘Process indicators’ section later in this chapter) such as bacterial spores, which are considered as the most resistant infectious agents (with the exception of prions, the agents responsible for spongiform encephalopathies). This is usually the situation for official sterilization methods. Pharmacopoeial recommendations as well as guideline documents are derived from data generated from the use of bacterial spores (biological indicators) for a given sterilization process.
When a fully validated sterilization process has been conducted, the release of a batch of product can be based on process data during sterilization rather than the results from sterility testing. Any change in the sterilization procedure (e.g. product load, type of containers) requires re-validation to take place.
Re-sterilization of products/devices can cause their degradation (e.g. repeated irradiation or autoclaving) or may even cause them to become toxic (e.g. with ethylene oxide; Richards 2004). Therefore any proposed re-sterilization must be carefully investigated.
Recommended pharmacopoeial sterilization processes
Five main sterilization processes which possess different characteristics are usually recommended by pharmacopoeias:
• steam (under pressure) sterilization (terminal)
• dry heat sterilization (terminal)
• ionizing radiation sterilization (terminal)
• gaseous (ethylene oxide) sterilization (terminal)
Although the use of other sterilization methodologies is not necessarily precluded, appropriate validation documentations for each product need to be provided. More information can be found in Chapters 15 and 16, or by consulting the relevant pharmacopoeia. At the time of writing, examples of these include the European Pharmacopoeia (2011), the United States Pharmacopeia (2012) and the British Pharmacopoeia (2013a) but it is always important to consult the most up-to-date texts and guidelines.
Steam (under pressure) sterilization
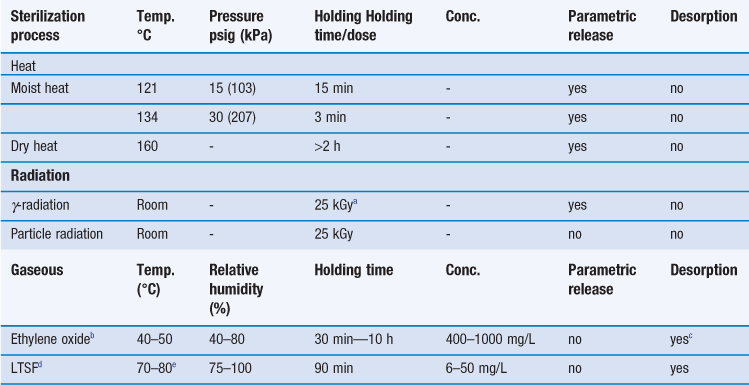
Steam sterilization is the most reliable, versatile and universally used form of sterilization and relies on the combination of steam, temperature and pressure. The typical cycle consists of a holding time of 15 minutes at a temperature of 121 °C under 15 psi (103 kPa) gauge pressure (Table 17.4). The aim is to deliver steam at the phase boundary (dry saturated steam; see Chapter 16, Fig. 16.2) to all areas of the load. This is achieved using steam and pressure (Table 17.5).
Table 17.4
Typical terminal sterilization cycles
cDesorption could take up to 15 days; maximum threshold of ethylene oxide residues and evaluation documented in ISO 10993–7 (1996).
dLow temperature steam formaldehyde; values can differ slightly depending upon the literature.
eLower temperature of 55–56 °C can be used depending upon the thermotolerance of the preparation.
Steam under pressure is commonly used unless prohibited by lack of load penetration or heat and/or moisture damage. Steam can only kill microorganisms if it makes direct contact, so it is very important to avoid air pockets in the sterilizer during a sterilization process. In addition, air can reduce the partial pressure of the steam so that the temperature reached on surfaces will be less than that expected with the pressure used. Hence, removal of air is an essential part of the process to ensure effective sterilization. To remove the air present when an autoclave is loaded, autoclaves are equipped with air removal/displacement systems (e.g. vacuum and displacement autoclaves, etc.). For porous loads, gravity displacement systems (downward-displacement autoclaves) are not adequate and vacuum and pressure-pulsing autoclaves are the method of choice (McDonnell 2007). Non-condensable gases must also be removed and monitored; these are atmospheric gases like nitrogen and oxygen that form part of the initial atmosphere of the sterilizer. Other factors that affect the efficacy of steam sterilization are water content and steam purity. The optimal sterilization is obtained with saturated steam (as discussed in Chapter 16). Supersaturated steam (i.e. wetter steam) is associated with condensation and poor penetration. Superheated steam (i.e. drier steam) behaves like dry heat and is less efficient. Steam purity is determined by the quality of the water, which can be affected by a number of contaminants (e.g. pyrogens, amines, toxic metals, iron, chlorides, etc.) that can render the sterile product unsafe (e.g. toxicity caused by pyrogenic reactions, metallic poisoning) or damaged (e.g. discoloration of packaging, corrosion caused by iron and chlorides).
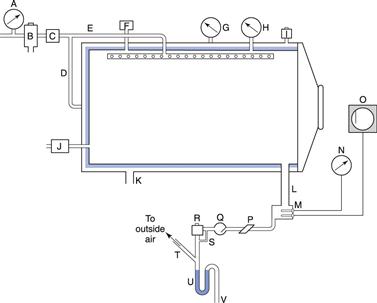
Steam under pressure is generated in autoclaves which can vary greatly in size and shape from portable bench-top units to industrial production facilities (Fig. 17.1). A cross-section through an autoclave is shown in Figure 17.2.



Steam sterilization applications are informed/regulated by a number of European and international guidelines and standards providing information on sterilizer design and installation, quality of steam, requirement for pressure, development and validation and routine control, etc.
Dry heat sterilization
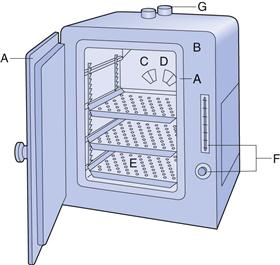
The most common dry heat sterilization method uses hot air ovens (Fig. 17.3). Other procedures, such as sterilizing tunnels utilizing high-temperature filtered laminar air flow or infrared irradiation to achieve rapid heat transfer, are also available. Hot air ovens are usually heated electrically and often have heaters under a perforated bottom plate to provide convection currents (gravity convection type). Mechanical convection hot air ovens are equipped with a fan to assist air circulation and increase heat transfer by convection (Joslyn 2001). Dry heat sterilization is less expensive than steam sterilization and is effective for the depyrogenation of containers/packaging (e.g. glassware). Overloading should be avoided, wrappings and other barriers minimized and the load positioned to allow optimal air circulation. Other problems include long heating up times (e.g. with large loads of instruments) and the charring or baking of organic matter onto items. Dry heat sterilization cycles are generally longer than for moist heat sterilization, typically 2 hours at 160 °C (see Table 17.4). The process is thermostatically controlled and monitored using thermocouples.
Dry heat sterilization cannot be used for a number of products such as rubber, plastics and other thermolabile items, or for aqueous solutions.
Integrated lethality in sterilization practice
All heat sterilization processes must include heating up and cooling down time periods. These prolonged time periods at a raised temperature may increase the degradation of the product. Integrated lethality attempts to examine the effects of heat on the inactivation process during these time periods.
For steam sterilization, the Fo concept (‘reference unit of lethality’) is used. This takes into account the heating up and cooling down stages of the cycle and is expressed as the equivalent time in minutes at a temperature of 121 °C delivered by the process to the product in its final container with reference to microorganisms possessing a Z value of 10. Its calculation is complex and further information can be found in the relevant pharmacopoeias. In practice, computer programs can be used to calculate the combined effect of whole processes, allowing a reduction in the total process time. It is important that the appropriate sterility assurance level is consistently achieved and the routine use of biological indicators is recommended, although following process validation, parametric release might be preferred.
Gaseous sterilization
The gaseous sterilization method recommended by pharmacopoeias mainly employs ethylene oxide. It is usually used on a commercial scale for the sterilization of catheters, infusion giving sets, syringes, prostheses and some plastic containers and thermolabile powders (if humidity is not a problem; Sharp 2000). The ethylene oxide sterilization cycle is complex since many factors need to be controlled over a long period of time (see Table 17.4
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


