FIGURE 39-5 Hollenhorst plaque lodged at the bifurcation of a retinal arteriole proves that a patient is shedding emboli from the carotid artery, great vessels, or heart.
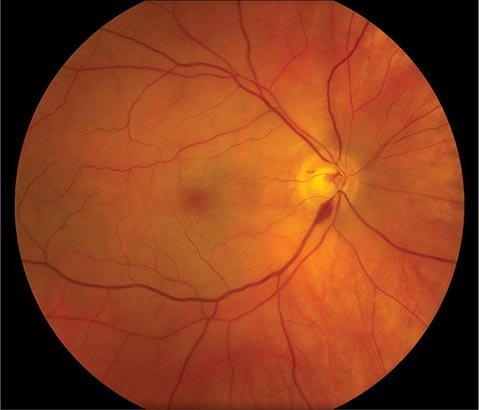
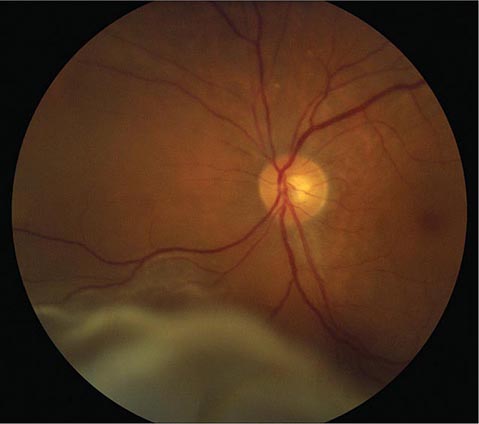
FIGURE 39-6 Central retinal artery occlusion in a 78-year-old man reducing acuity to counting fingers in the right eye. Note the splinter hemorrhage on the optic disc and the slightly milky appearance to the macula with a cherry-red fovea.
In rare instances, amaurosis fugax results from low central retinal artery perfusion pressure in a patient with a critical stenosis of the ipsilateral carotid artery and poor collateral flow via the circle of Willis. In this situation, amaurosis fugax develops when there is a dip in systemic blood pressure or a slight worsening of the carotid stenosis. Sometimes there is contralateral motor or sensory loss, indicating concomitant hemispheric cerebral ischemia.
Retinal arterial occlusion also occurs rarely in association with retinal migraine, lupus erythematosus, anticardiolipin antibodies, anticoagulant deficiency states (protein S, protein C, and antithrombin deficiency), pregnancy, IV drug abuse, blood dyscrasias, dysproteinemias, and temporal arteritis.
Marked systemic hypertension causes sclerosis of retinal arterioles, splinter hemorrhages, focal infarcts of the nerve fiber layer (cotton-wool spots), and leakage of lipid and fluid (hard exudate) into the macula (Fig. 39-7). In hypertensive crisis, sudden visual loss can result from vasospasm of retinal arterioles and retinal ischemia. In addition, acute hypertension may produce visual loss from ischemic swelling of the optic disc. Patients with acute hypertensive retinopathy should be treated by lowering the blood pressure. However, the blood pressure should not be reduced precipitously, because there is a danger of optic disc infarction from sudden hypoperfusion.
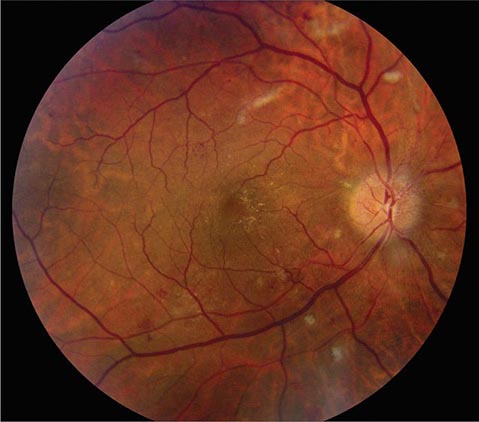
FIGURE 39-7 Hypertensive retinopathy with blurred optic disc, scattered hemorrhages, cotton-wool spots (nerve fiber layer infarcts), and foveal exudate in a 62-year-old man with chronic renal failure and a systolic blood pressure of 220.
Impending branch or central retinal vein occlusion can produce prolonged visual obscurations that resemble those described by patients with amaurosis fugax. The veins appear engorged and phlebitic, with numerous retinal hemorrhages (Fig. 39-8). In some patients, venous blood flow recovers spontaneously, whereas others evolve a frank obstruction with extensive retinal bleeding (“blood and thunder” appearance), infarction, and visual loss. Venous occlusion of the retina is often idiopathic, but hypertension, diabetes, and glaucoma are prominent risk factors. Polycythemia, thrombocythemia, or other factors leading to an underlying hypercoagulable state should be corrected; aspirin treatment may be beneficial.
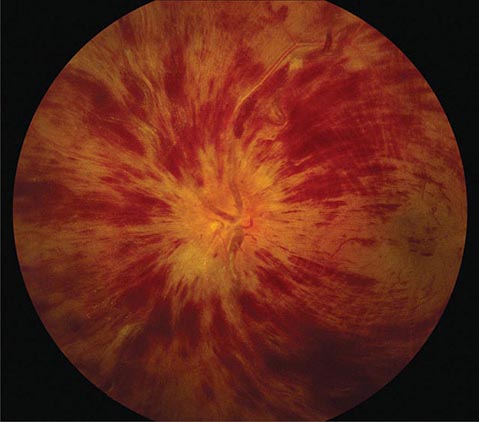
FIGURE 39-8 Central retinal vein occlusion can produce massive retinal hemorrhage (“blood and thunder”), ischemia, and vision loss.
Anterior Ischemic Optic Neuropathy (AION) This is caused by insufficient blood flow through the posterior ciliary arteries that supply the optic disc. It produces painless monocular visual loss that is sudden in onset, followed sometimes by stuttering progression. The optic disc appears swollen and surrounded by nerve fiber layer splinter hemorrhages (Fig. 39-9). AION is divided into two forms: arteritic and nonarteritic. The nonarteritic form is most common. No specific cause can be identified, although diabetes and hypertension are common risk factors. A crowded disc architecture and small optic cup predispose to the development of nonarteritic AION. No treatment is available. About 5% of patients, especially those age >60, develop the arteritic form of AION in conjunction with giant-cell (temporal) arteritis (Chap. 385). It is urgent to recognize arteritic AION so that high doses of glucocorticoids can be instituted immediately to prevent blindness in the second eye. Symptoms of polymyalgia rheumatica may be present; the sedimentation rate and C-reactive protein level are usually elevated. In a patient with visual loss from suspected arteritic AION, temporal artery biopsy is mandatory to confirm the diagnosis. Glucocorticoids should be started immediately, without waiting for the biopsy to be completed. The diagnosis of arteritic AION is difficult to sustain in the face of a negative temporal artery biopsy, but such cases do occur rarely. It is important to biopsy an arterial segment of at least 3 cm and to examine a sufficient number of tissue sections prepared from the specimen.
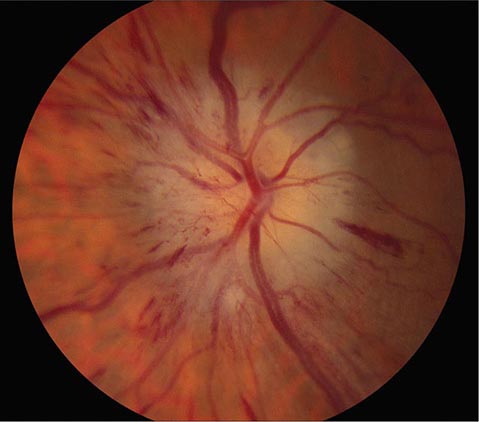
FIGURE 39-9 Anterior ischemic optic neuropathy from temporal arteritis in a 67-year-old woman with acute disc swelling, splinter hemorrhages, visual loss, and an erythrocyte sedimentation rate of 70 mm/h.
Posterior Ischemic Optic Neuropathy This is an uncommon cause of acute visual loss, induced by the combination of severe anemia and hypotension. Cases have been reported after major blood loss during surgery (especially in patients undergoing cardiac or lumbar spine operations), exsanguinating trauma, gastrointestinal bleeding, and renal dialysis. The fundus usually appears normal, although optic disc swelling develops if the process extends anteriorly far enough to reach the globe. Vision can be salvaged in some patients by prompt blood transfusion and reversal of hypotension.
Optic Neuritis This is a common inflammatory disease of the optic nerve. In the Optic Neuritis Treatment Trial (ONTT), the mean age of patients was 32 years, 77% were female, 92% had ocular pain (especially with eye movements), and 35% had optic disc swelling. In most patients, the demyelinating event was retrobulbar and the ocular fundus appeared normal on initial examination (Fig. 39-10), although optic disc pallor slowly developed over subsequent months.
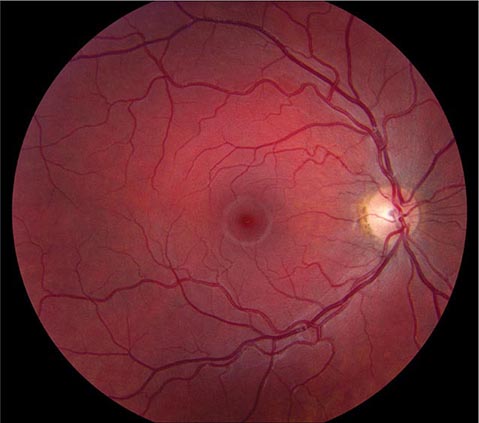
FIGURE 39-10 Retrobulbar optic neuritis is characterized by a normal fundus examination initially, hence the rubric “the doctor sees nothing, and the patient sees nothing.” Optic atrophy develops after severe or repeated attacks.
Virtually all patients experience a gradual recovery of vision after a single episode of optic neuritis, even without treatment. This rule is so reliable that failure of vision to improve after a first attack of optic neuritis casts doubt on the original diagnosis. Treatment with high-dose IV methylprednisolone (250 mg every 6 h for 3 days) followed by oral prednisone (1 mg/kg per day for 11 days) makes no difference in ultimate acuity 6 months after the attack, but the recovery of visual function occurs more rapidly. Therefore, when visual loss is severe (worse than 20/100), IV followed by PO glucocorticoids are often recommended.
For some patients, optic neuritis remains an isolated event. However, the ONTT showed that the 15-year cumulative probability of developing clinically definite multiple sclerosis after optic neuritis is 50%. A brain magnetic resonance (MR) scan is advisable in every patient with a first attack of optic neuritis. If two or more plaques are present on initial imaging, treatment should be considered to prevent the development of additional demyelinating lesions (Chap. 458).
LEBER’S HEREDITARY OPTIC NEUROPATHY
This disease usually affects young men, causing gradual, painless, severe central visual loss in one eye, followed weeks to years later by the same process in the other eye. Acutely, the optic disc appears mildly plethoric with surface capillary telangiectasias but no vascular leakage on fluorescein angiography. Eventually optic atrophy ensues. Leber’s optic neuropathy is caused by a point mutation at codon 11778 in the mitochondrial gene encoding nicotinamide adenine dinucleotide dehydrogenase (NADH) subunit 4. Additional mutations responsible for the disease have been identified, most in mitochondrial genes that encode proteins involved in electron transport. Mitochondrial mutations that cause Leber’s neuropathy are inherited from the mother by all her children, but usually only sons develop symptoms.
Toxic Optic Neuropathy This can result in acute visual loss with bilateral optic disc swelling and central or cecocentral scotomas. Such cases have been reported to result from exposure to ethambutol, methyl alcohol (moonshine), ethylene glycol (antifreeze), or carbon monoxide. In toxic optic neuropathy, visual loss also can develop gradually and produce optic atrophy (Fig. 39-11) without a phase of acute optic disc edema. Many agents have been implicated as a cause of toxic optic neuropathy, but the evidence supporting the association for many is weak. The following is a partial list of potential offending drugs or toxins: disulfiram, ethchlorvynol, chloramphenicol, amiodarone, monoclonal anti-CD3 antibody, ciprofloxacin, digitalis, streptomycin, lead, arsenic, thallium, D-penicillamine, isoniazid, emetine, sildenafil, tadalafil, vardenafil, and sulfonamides. Deficiency states induced by starvation, malabsorption, or alcoholism can lead to insidious visual loss. Thiamine, vitamin B12, and folate levels should be checked in any patient with unexplained bilateral central scotomas and optic pallor.
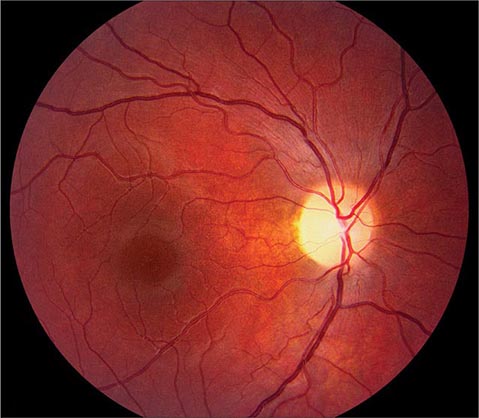
FIGURE 39-11 Optic atrophy is not a specific diagnosis but refers to the combination of optic disc pallor, arteriolar narrowing, and nerve fiber layer destruction produced by a host of eye diseases, especially optic neuropathies.
Papilledema This connotes bilateral optic disc swelling from raised intracranial pressure (Fig. 39-12). Headache is a common but not invariable accompaniment. All other forms of optic disc swelling (e.g., from optic neuritis or ischemic optic neuropathy) should be called “optic disc edema”. This convention is arbitrary but serves to avoid confusion. Often it is difficult to differentiate papilledema from other forms of optic disc edema by fundus examination alone. Transient visual obscurations are a classic symptom of papilledema. They can occur in only one eye or simultaneously in both eyes. They usually last seconds but can persist longer. Obscurations follow abrupt shifts in posture or happen spontaneously. When obscurations are prolonged or spontaneous, the papilledema is more threatening. Visual acuity is not affected by papilledema unless the papilledema is severe, long-standing, or accompanied by macular edema and hemorrhage. Visual field testing shows enlarged blind spots and peripheral constriction (Fig. 39-3F). With unremitting papilledema, peripheral visual field loss progresses in an insidious fashion while the optic nerve develops atrophy. In this setting, reduction of optic disc swelling is an ominous sign of a dying nerve rather than an encouraging indication of resolving papilledema.
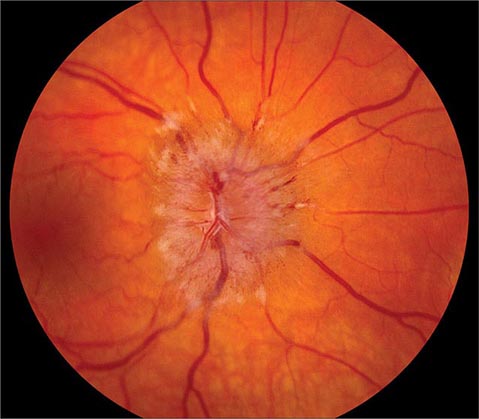
FIGURE 39-12 Papilledema means optic disc edema from raised intracranial pressure. This young woman developed acute papilledema, with hemorrhages and cotton-wool spots, as a rare side effect of treatment with tetracycline for acne.
Evaluation of papilledema requires neuroimaging to exclude an intracranial lesion. MR angiography is appropriate in selected cases to search for a dural venous sinus occlusion or an arteriovenous shunt. If neuroradiologic studies are negative, the subarachnoid opening pressure should be measured by lumbar puncture. An elevated pressure, with normal cerebrospinal fluid, points by exclusion to the diagnosis of pseudotumor cerebri (idiopathic intracranial hypertension). The majority of patients are young, female, and obese. Treatment with a carbonic anhydrase inhibitor such as acetazolamide lowers intracranial pressure by reducing the production of cerebrospinal fluid. Weight reduction is vital: bariatric surgery should be considered in patients who cannot lose weight by diet control. If vision loss is severe or progressive, a shunt should be performed without delay to prevent blindness. Occasionally, emergency surgery is required for sudden blindness caused by fulminant papilledema.
Optic Disc Drusen These are refractile deposits within the substance of the optic nerve head (Fig. 39-13). They are unrelated to drusen of the retina, which occur in age-related macular degeneration. Optic disc drusen are most common in people of northern European descent. Their diagnosis is obvious when they are visible as glittering particles on the surface of the optic disc. However, in many patients they are hidden beneath the surface, producing pseudopapilledema. It is important to recognize optic disc drusen to avoid an unnecessary evaluation for papilledema. Ultrasound or computed tomography (CT) scanning is sensitive for detection of buried optic disc drusen because they contain calcium. In most patients, optic disc drusen are an incidental, innocuous finding, but they can produce visual obscurations. On perimetry they give rise to enlarged blind spots and arcuate scotomas from damage to the optic disc. With increasing age, drusen tend to become more exposed on the disc surface as optic atrophy develops. Hemorrhage, choroidal neovascular membrane, and AION are more likely to occur in patients with optic disc drusen. No treatment is available.
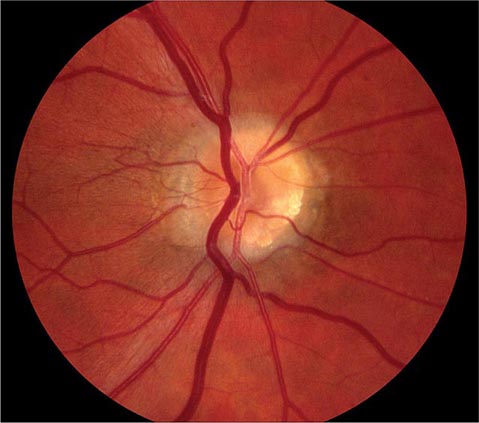
FIGURE 39-13 Optic disc drusen are calcified, mulberry-like deposits of unknown etiology within the optic disc, giving rise to “pseudopapilledema.”
Vitreous Degeneration This occurs in all individuals with advancing age, leading to visual symptoms. Opacities develop in the vitreous, casting annoying shadows on the retina. As the eye moves, these distracting “floaters” move synchronously, with a slight lag caused by inertia of the vitreous gel. Vitreous traction on the retina causes mechanical stimulation, resulting in perception of flashing lights. This photopsia is brief and is confined to one eye, in contrast to the bilateral, prolonged scintillations of cortical migraine. Contraction of the vitreous can result in sudden separation from the retina, heralded by an alarming shower of floaters and photopsia. This process, known as vitreous detachment, is a common involutional event in the elderly. It is not harmful unless it damages the retina. A careful examination of the dilated fundus is important in any patient complaining of floaters or photopsia to search for peripheral tears or holes. If such a lesion is found, laser application can forestall a retinal detachment. Occasionally a tear ruptures a retinal blood vessel, causing vitreous hemorrhage and sudden loss of vision. On attempted ophthalmoscopy the fundus is hidden by a dark haze of blood. Ultrasound is required to examine the interior of the eye for a retinal tear or detachment. If the hemorrhage does not resolve spontaneously, the vitreous can be removed surgically. Vitreous hemorrhage also results from the fragile neovascular vessels that proliferate on the surface of the retina in diabetes, sickle cell anemia, and other ischemic ocular diseases.
Retinal Detachment This produces symptoms of floaters, flashing lights, and a scotoma in the peripheral visual field corresponding to the detachment (Fig. 39-14). If the detachment includes the fovea, there is an afferent pupil defect and the visual acuity is reduced. In most eyes, retinal detachment starts with a hole, flap, or tear in the peripheral retina (rhegmatogenous retinal detachment). Patients with peripheral retinal thinning (lattice degeneration) are particularly vulnerable to this process. Once a break has developed in the retina, liquefied vitreous is free to enter the subretinal space, separating the retina from the pigment epithelium. The combination of vitreous traction on the retinal surface and passage of fluid behind the retina leads inexorably to detachment. Patients with a history of myopia, trauma, or prior cataract extraction are at greatest risk for retinal detachment. The diagnosis is confirmed by ophthalmoscopic examination of the dilated eye.
Classic Migraine (See also Chap. 447) This usually occurs with a visual aura lasting about 20 min. In a typical attack, a small central disturbance in the field of vision marches toward the periphery, leaving a transient scotoma in its wake. The expanding border of migraine scotoma has a scintillating, dancing, or zigzag edge, resembling the bastions of a fortified city, hence the term fortification spectra. Patients’ descriptions of fortification spectra vary widely and can be confused with amaurosis fugax. Migraine patterns usually last longer and are perceived in both eyes, whereas amaurosis fugax is briefer and occurs in only one eye. Migraine phenomena also remain visible in the dark or with the eyes closed. Generally they are confined to either the right or the left visual hemifield, but sometimes both fields are involved simultaneously. Patients often have a long history of stereotypic attacks. After the visual symptoms recede, headache develops in most patients.
FIGURE 39-14 Retinal detachment appears as an elevated sheet of retinal tissue with folds. In this patient, the fovea was spared, so acuity was normal, but an inferior detachment produced a superior scotoma.
Transient Ischemic Attacks Vertebrobasilar insufficiency may result in acute homonymous visual symptoms. Many patients mistakenly describe symptoms in the left or right eye when in fact the symptoms are occurring in the left or right hemifield of both eyes. Interruption of blood supply to the visual cortex causes a sudden fogging or graying of vision, occasionally with flashing lights or other positive phenomena that mimic migraine. Cortical ischemic attacks are briefer in duration than migraine, occur in older patients, and are not followed by headache. There may be associated signs of brainstem ischemia, such as diplopia, vertigo, numbness, weakness, and dysarthria.
Stroke Stroke occurs when interruption of blood supply from the posterior cerebral artery to the visual cortex is prolonged. The only finding on examination is a homonymous visual field defect that stops abruptly at the vertical meridian. Occipital lobe stroke usually is due to thrombotic occlusion of the vertebrobasilar system, embolus, or dissection. Lobar hemorrhage, tumor, abscess, and arteriovenous malformation are other common causes of hemianopic cortical visual loss.
Factitious (Functional, Nonorganic) Visual Loss This is claimed by hysterics or malingerers. The latter account for the vast majority, seeking sympathy, special treatment, or financial gain by feigning loss of sight. The diagnosis is suspected when the history is atypical, physical findings are lacking or contradictory, inconsistencies emerge on testing, and a secondary motive can be identified. In our litigious society, the fraudulent pursuit of recompense has spawned an epidemic of factitious visual loss.
CHRONIC VISUAL LOSS
Cataract Cataract is a clouding of the lens sufficient to reduce vision. Most cataracts develop slowly as a result of aging, leading to gradual impairment of vision. The formation of cataract occurs more rapidly in patients with a history of ocular trauma, uveitis, or diabetes mellitus. Cataracts are acquired in a variety of genetic diseases, such as myotonic dystrophy, neurofibromatosis type 2, and galactosemia. Radiation therapy and glucocorticoid treatment can induce cataract as a side effect. The cataracts associated with radiation or glucocorticoids have a typical posterior subcapsular location. Cataract can be detected by noting an impaired red reflex when viewing light reflected from the fundus with an ophthalmoscope or by examining the dilated eye with the slit lamp.
The only treatment for cataract is surgical extraction of the opacified lens. Millions of cataract operations are performed each year around the globe. The operation generally is done under local anesthesia on an outpatient basis. A plastic or silicone intraocular lens is placed within the empty lens capsule in the posterior chamber, substituting for the natural lens and leading to rapid recovery of sight. More than 95% of patients who undergo cataract extraction can expect an improvement in vision. In some patients, the lens capsule remaining in the eye after cataract extraction eventually turns cloudy, causing secondary loss of vision. A small opening, called a posterior capsulotomy, is made in the lens capsule with a laser to restore clarity.
Glaucoma Glaucoma is a slowly progressive, insidious optic neuropathy that usually is associated with chronic elevation of intraocular pressure. After cataract, it is the most common cause of blindness in the world. It is especially prevalent in people of African descent. The mechanism by which raised intraocular pressure injures the optic nerve is not understood. Axons entering the inferotemporal and superotemporal aspects of the optic disc are damaged first, producing typical nerve fiber bundle or arcuate scotomas on perimetric testing. As fibers are destroyed, the neural rim of the optic disc shrinks and the physiologic cup within the optic disc enlarges (Fig. 39-15). This process is referred to as pathologic “cupping.” The cup-to-disc diameter is expressed as a fraction (e.g., 0.2). The cup-to-disc ratio ranges widely in normal individuals, making it difficult to diagnose glaucoma reliably simply by observing an unusually large or deep optic cup. Careful documentation of serial examinations is helpful. In a patient with physiologic cupping the large cup remains stable, whereas in a patient with glaucoma it expands relentlessly over the years. Observation of progressive cupping and detection of an arcuate scotoma or a nasal step on computerized visual field testing is sufficient to establish the diagnosis of glaucoma. Optical coherence tomography reveals corresponding loss of fibers along the arcuate pathways in the nerve fiber layer.
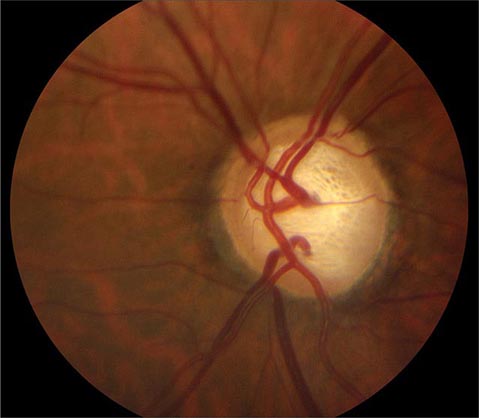
FIGURE 39-15 Glaucoma results in “cupping” as the neural rim is destroyed and the central cup becomes enlarged and excavated. The cup-to-disc ratio is about 0.8 in this patient.
About 95% of patients with glaucoma have open anterior chamber angles. In most affected individuals the intraocular pressure is elevated. The cause of elevated intraocular pressure is unknown, but it is associated with gene mutations in the heritable forms. Surprisingly, a third of patients with open-angle glaucoma have an intraocular pressure within the normal range of 10–20 mmHg. For this so-called normal or low-tension form of glaucoma, high myopia is a risk factor.
Chronic angle-closure glaucoma and chronic open-angle glaucoma are usually asymptomatic. Only acute angle-closure glaucoma causes a red or painful eye, from abrupt elevation of intraocular pressure. In all forms of glaucoma, foveal acuity is spared until end-stage disease is reached. For these reasons, severe and irreversible damage can occur before either the patient or the physician recognizes the diagnosis. Screening of patients for glaucoma by noting the cup-to-disc ratio on ophthalmoscopy and by measuring intraocular pressure is vital. Glaucoma is treated with topical adrenergic agonists, cholinergic agonists, beta blockers, and prostaglandin analogues. Occasionally, systemic absorption of beta blocker from eyedrops can be sufficient to cause side effects of bradycardia, hypotension, heart block, bronchospasm, or depression. Topical or oral carbonic anhydrase inhibitors are used to lower intraocular pressure by reducing aqueous production. Laser treatment of the trabecular meshwork in the anterior chamber angle improves aqueous outflow from the eye. If medical or laser treatments fail to halt optic nerve damage from glaucoma, a filter must be constructed surgically (trabeculectomy) or a drainage device placed to release aqueous from the eye in a controlled fashion.
Macular Degeneration This is a major cause of gradual, painless, bilateral central visual loss in the elderly. It occurs in a nonexudative (dry) form and an exudative (wet) form. Inflammation may be important in both forms of macular degeneration; susceptibility is associated with variants in the gene for complement factor H, an inhibitor of the alternative complement pathway. The nonexudative process begins with the accumulation of extracellular deposits called drusen underneath the retinal pigment epithelium. On ophthalmoscopy, they are pleomorphic but generally appear as small discrete yellow lesions clustered in the macula (Fig. 39-16). With time they become larger, more numerous, and confluent. The retinal pigment epithelium becomes focally detached and atrophic, causing visual loss by interfering with photoreceptor function. Treatment with vitamins C and E, beta-carotene, and zinc may retard dry macular degeneration.
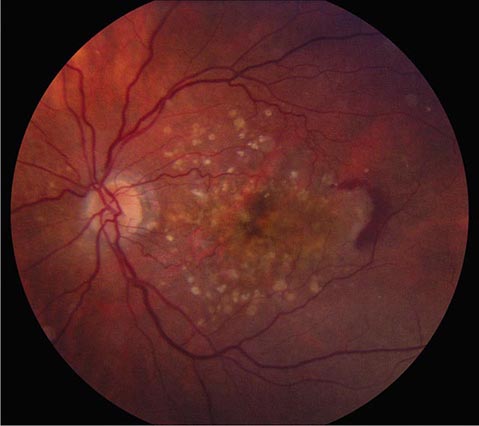
FIGURE 39-16 Age-related macular degeneration consisting of scattered yellow drusen in the macula (dry form) and a crescent of fresh hemorrhage temporal to the fovea from a subretinal neovascular membrane (wet form).
Exudative macular degeneration, which develops in only a minority of patients, occurs when neovascular vessels from the choroid grow through defects in Bruch’s membrane and proliferate underneath the retinal pigment epithelium or the retina. Leakage from these vessels produces elevation of the retina, with distortion (metamorphopsia) and blurring of vision. Although the onset of these symptoms is usually gradual, bleeding from a subretinal choroidal neovascular membrane sometimes causes acute visual loss. Neovascular membranes can be difficult to see on fundus examination because they are located beneath the retina. Fluorescein angiography and optical coherence tomography, a technique for acquiring images of the retina in cross-section, are extremely useful for their detection. Major or repeated hemorrhage under the retina from neovascular membranes results in fibrosis, development of a round (disciform) macular scar, and permanent loss of central vision.
A major therapeutic advance has occurred with the discovery that exudative macular degeneration can be treated with intraocular injection of antagonists to vascular endothelial growth factor. Bevacizumab, ranibizumab, or aflibercept is administered by direct injection into the vitreous cavity, beginning on a monthly basis. These antibodies cause the regression of neovascular membranes by blocking the action of vascular endothelial growth factor, thereby improving visual acuity.
Central Serous Chorioretinopathy This primarily affects males between the ages of 20 and 50 years. Leakage of serous fluid from the choroid causes small, localized detachment of the retinal pigment epithelium and the neurosensory retina. These detachments produce acute or chronic symptoms of metamorphopsia and blurred vision when the macula is involved. They are difficult to visualize with a direct ophthalmoscope because the detached retina is transparent and only slightly elevated. Optical coherence tomography shows fluid beneath the retina, and fluorescein angiography shows dye streaming into the subretinal space. The cause of central serous chorioretinopathy is unknown. Symptoms may resolve spontaneously if the retina reattaches, but recurrent detachment is common. Laser photocoagulation has benefited some patients with this condition.
Diabetic Retinopathy A rare disease until 1921, when the discovery of insulin resulted in a dramatic improvement in life expectancy for patients with diabetes mellitus, diabetic retinopathy is now a leading cause of blindness in the United States. The retinopathy takes years to develop but eventually appears in nearly all cases. Regular surveillance of the dilated fundus is crucial for any patient with diabetes. In advanced diabetic retinopathy, the proliferation of neovascular vessels leads to blindness from vitreous hemorrhage, retinal detachment, and glaucoma (Fig. 39-17). These complications can be avoided in most patients by administration of panretinal laser photocoagulation at the appropriate point in the evolution of the disease. For further discussion of the manifestations and management of diabetic retinopathy, see Chaps. 417–419.
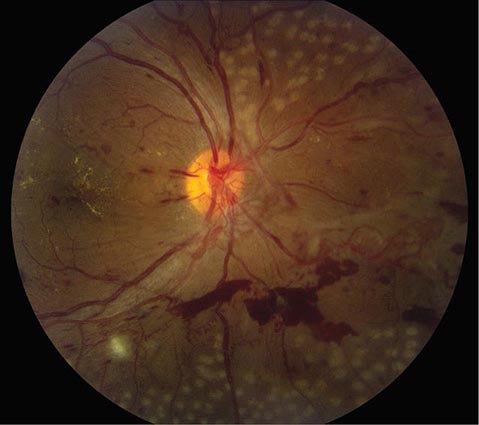
FIGURE 39-17 Proliferative diabetic retinopathy in a 25-year-old man with an 18-year history of diabetes, showing neovascular vessels emanating from the optic disc, retinal and vitreous hemorrhage, cotton-wool spots, and macular exudate. Round spots in the periphery represent recently applied panretinal photocoagulation.
Retinitis Pigmentosa This is a general term for a disparate group of rod-cone dystrophies characterized by progressive night blindness, visual field constriction with a ring scotoma, loss of acuity, and an abnormal electroretinogram (ERG). It occurs sporadically or in an autosomal recessive, dominant, or X-linked pattern. Irregular black deposits of clumped pigment in the peripheral retina, called bone spicules because of their vague resemblance to the spicules of cancellous bone, give the disease its name (Fig. 39-18). The name is actually a misnomer because retinitis pigmentosa is not an inflammatory process. Most cases are due to a mutation in the gene for rhodopsin, the rod photopigment, or in the gene for peripherin, a glycoprotein located in photoreceptor outer segments. Vitamin A (15,000 IU/d) slightly retards the deterioration of the ERG in patients with retinitis pigmentosa but has no beneficial effect on visual acuity or fields.
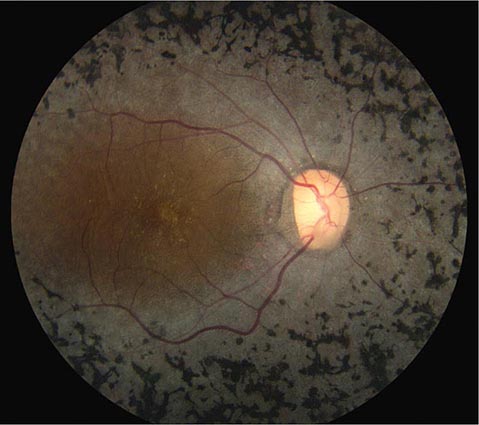
FIGURE 39-18 Retinitis pigmentosa with black clumps of pigment known as “bone spicules.” The patient had peripheral visual field loss with sparing of central (macular) vision.
Leber’s congenital amaurosis, a rare cone dystrophy, has been treated by replacement of the missing RPE65 protein through gene therapy, resulting in modest improvement in visual function. Some forms of retinitis pigmentosa occur in association with rare, hereditary systemic diseases (olivopontocerebellar degeneration, Bassen-Kornzweig disease, Kearns-Sayre syndrome, Refsum’s disease). Chronic treatment with chloroquine, hydroxychloroquine, and phenothiazines (especially thioridazine) can produce visual loss from a toxic retinopathy that resembles retinitis pigmentosa.
Epiretinal Membrane This is a fibrocellular tissue that grows across the inner surface of the retina, causing metamorphopsia and reduced visual acuity from distortion of the macula. A crinkled, cellophane-like membrane is visible on the retinal examination. Epiretinal membrane is most common in patients over 50 years of age and is usually unilateral. Most cases are idiopathic, but some occur as a result of hypertensive retinopathy, diabetes, retinal detachment, or trauma. When visual acuity is reduced to the level of about 6/24 (20/80), vitrectomy and surgical peeling of the membrane to relieve macular puckering are recommended. Contraction of an epiretinal membrane sometimes gives rise to a macular hole. Most macular holes, however, are caused by local vitreous traction within the fovea. Vitrectomy can improve acuity in selected cases.
Melanoma and Other Tumors Melanoma is the most common primary tumor of the eye (Fig. 39-19). It causes photopsia, an enlarging scotoma, and loss of vision. A small melanoma is often difficult to differentiate from a benign choroidal nevus. Serial examinations are required to document a malignant pattern of growth. Treatment of melanoma is controversial. Options include enucleation, local resection, and irradiation. Metastatic tumors to the eye outnumber primary tumors. Breast and lung carcinomas have a special propensity to spread to the choroid or iris. Leukemia and lymphoma also commonly invade ocular tissues. Sometimes their only sign on eye examination is cellular debris in the vitreous, which can masquerade as a chronic posterior uveitis. Retrobulbar tumor of the optic nerve (meningioma, glioma) or chiasmal tumor (pituitary adenoma, meningioma) produces gradual visual loss with few objective findings except for optic disc pallor. Rarely, sudden expansion of a pituitary adenoma from infarction and bleeding (pituitary apoplexy) causes acute retrobulbar visual loss, with headache, nausea, and ocular motor nerve palsies. In any patient with visual field loss or optic atrophy, CT or MR scanning should be considered if the cause remains unknown after careful review of the history and thorough examination of the eye.
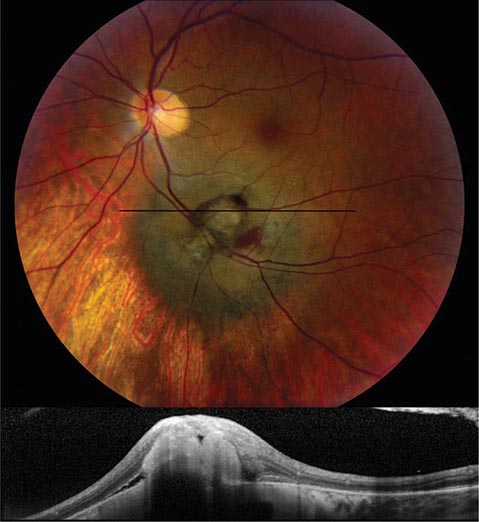
FIGURE 39-19 Melanoma of the choroid, appearing as an elevated dark mass in the inferior fundus, with overlying hemorrhage. The black line denotes the plane of the optical coherence tomography scan (below) showing the subretinal tumor.
PROPTOSIS
When the globes appear asymmetric, the clinician must first decide which eye is abnormal. Is one eye recessed within the orbit (enophthalmos), or is the other eye protuberant (exophthalmos, or proptosis)? A small globe or a Horner’s syndrome can give the appearance of enophthalmos. True enophthalmos occurs commonly after trauma, from atrophy of retrobulbar fat, or from fracture of the orbital floor. The position of the eyes within the orbits is measured by using a Hertel exophthalmometer, a handheld instrument that records the position of the anterior corneal surface relative to the lateral orbital rim. If this instrument is not available, relative eye position can be judged by bending the patient’s head forward and looking down upon the orbits. A proptosis of only 2 mm in one eye is detectable from this perspective. The development of proptosis implies a space-occupying lesion in the orbit and usually warrants CT or MR imaging.
Graves’ Ophthalmopathy This is the leading cause of proptosis in adults (Chap. 405). The proptosis is often asymmetric and can even appear to be unilateral. Orbital inflammation and engorgement of the extraocular muscles, particularly the medial rectus and the inferior rectus, account for the protrusion of the globe. Corneal exposure, lid retraction, conjunctival injection, restriction of gaze, diplopia, and visual loss from optic nerve compression are cardinal symptoms. Graves’ eye disease is a clinical diagnosis, but laboratory testing can be useful. The serum level of thyroid-stimulating immunoglobulins is often elevated. Orbital imaging usually reveals enlarged extraocular eye muscles, but not always. Graves’ ophthalmopathy can be treated with oral prednisone (60 mg/d) for 1 month, followed by a taper over several months. Worsening of symptoms upon glucocorticoid withdrawal is common. Topical lubricants, taping the eyelids closed at night, moisture chambers, and eyelid surgery are helpful to limit exposure of ocular tissues. Radiation therapy is not effective. Orbital decompression should be performed for severe, symptomatic exophthalmos or if visual function is reduced by optic nerve compression. In patients with diplopia, prisms or eye muscle surgery can be used to restore ocular alignment in primary gaze.
Orbital Pseudotumor This is an idiopathic, inflammatory orbital syndrome that is distinguished from Graves’ ophthalmopathy by the prominent complaint of pain. Other symptoms include diplopia, ptosis, proptosis, and orbital congestion. Evaluation for sarcoidosis, granulomatosis with polyangiitis, and other types of orbital vasculitis or collagen-vascular disease is negative. Imaging often shows swollen eye muscles (orbital myositis) with enlarged tendons. By contrast, in Graves’ ophthalmopathy, the tendons of the eye muscles usually are spared. The Tolosa-Hunt syndrome (Chap. 455) may be regarded as an extension of orbital pseudotumor through the superior orbital fissure into the cavernous sinus. The diagnosis of orbital pseudotumor is difficult. Biopsy of the orbit frequently yields nonspecific evidence of fat infiltration by lymphocytes, plasma cells, and eosinophils. A dramatic response to a therapeutic trial of systemic glucocorticoids indirectly provides the best confirmation of the diagnosis.
Orbital Cellulitis This causes pain, lid erythema, proptosis, conjunctival chemosis, restricted motility, decreased acuity, afferent pupillary defect, fever, and leukocytosis. It often arises from the paranasal sinuses, especially by contiguous spread of infection from the ethmoid sinus through the lamina papyracea of the medial orbit. A history of recent upper respiratory tract infection, chronic sinusitis, thick mucus secretions, or dental disease is significant in any patient with suspected orbital cellulitis. Blood cultures should be obtained, but they are usually negative. Most patients respond to empirical therapy with broad-spectrum IV antibiotics. Occasionally, orbital cellulitis follows an overwhelming course, with massive proptosis, blindness, septic cavernous sinus thrombosis, and meningitis. To avert this disaster, orbital cellulitis should be managed aggressively in the early stages, with immediate imaging of the orbits and antibiotic therapy that includes coverage of methicillin-resistant Staphylococcus aureus (MRSA). Prompt surgical drainage of an orbital abscess or paranasal sinusitis is indicated if optic nerve function deteriorates despite antibiotics.
Tumors Tumors of the orbit cause painless, progressive proptosis. The most common primary tumors are cavernous hemangioma, lymphangioma, neurofibroma, schwannoma, dermoid cyst, adenoid cystic carcinoma, optic nerve glioma, optic nerve meningioma, and benign mixed tumor of the lacrimal gland. Metastatic tumor to the orbit occurs frequently in breast carcinoma, lung carcinoma, and lymphoma. Diagnosis by fine-needle aspiration followed by urgent radiation therapy sometimes can preserve vision.
Carotid Cavernous Fistulas With anterior drainage through the orbit, these fistulas produce proptosis, diplopia, glaucoma, and corkscrew, arterialized conjunctival vessels. Direct fistulas usually result from trauma. They are easily diagnosed because of the prominent signs produced by high-flow, high-pressure shunting. Indirect fistulas, or dural arteriovenous malformations, are more likely to occur spontaneously, especially in older women. The signs are more subtle, and the diagnosis frequently is missed. The combination of slight proptosis, diplopia, enlarged muscles, and an injected eye often is mistaken for thyroid ophthalmopathy. A bruit heard upon auscultation of the head or reported by the patient is a valuable diagnostic clue. Imaging shows an enlarged superior ophthalmic vein in the orbits. Carotid cavernous shunts can be eliminated by intravascular embolization.
PTOSIS
Blepharoptosis This is an abnormal drooping of the eyelid. Unilateral or bilateral ptosis can be congenital, from dysgenesis of the levator palpebrae superioris, or from abnormal insertion of its aponeurosis into the eyelid. Acquired ptosis can develop so gradually that the patient is unaware of the problem. Inspection of old photographs is helpful in dating the onset. A history of prior trauma, eye surgery, contact lens use, diplopia, systemic symptoms (e.g., dysphagia or peripheral muscle weakness), or a family history of ptosis should be sought. Fluctuating ptosis that worsens late in the day is typical of myasthenia gravis. Examination should focus on evidence for proptosis, eyelid masses or deformities, inflammation, pupil inequality, or limitation of motility. The width of the palpebral fissures is measured in primary gaze to determine the degree of ptosis. The ptosis will be underestimated if the patient compensates by lifting the brow with the frontalis muscle.
Mechanical Ptosis This occurs in many elderly patients from stretching and redundancy of eyelid skin and subcutaneous fat (dermatochalasis). The extra weight of these sagging tissues causes the lid to droop. Enlargement or deformation of the eyelid from infection, tumor, trauma, or inflammation also results in ptosis on a purely mechanical basis.
Aponeurotic Ptosis This is an acquired dehiscence or stretching of the aponeurotic tendon, which connects the levator muscle to the tarsal plate of the eyelid. It occurs commonly in older patients, presumably from loss of connective tissue elasticity. Aponeurotic ptosis is also a common sequela of eyelid swelling from infection or blunt trauma to the orbit, cataract surgery, or contact lens use.
Myogenic Ptosis The causes of myogenic ptosis include myasthenia gravis (Chap. 461) and a number of rare myopathies that manifest with ptosis. The term chronic progressive external ophthalmoplegia refers to a spectrum of systemic diseases caused by mutations of mitochondrial DNA. As the name implies, the most prominent findings are symmetric, slowly progressive ptosis and limitation of eye movements. In general, diplopia is a late symptom because all eye movements are reduced equally. In the Kearns-Sayre variant, retinal pigmentary changes and abnormalities of cardiac conduction develop. Peripheral muscle biopsy shows characteristic “ragged-red fibers.” Oculopharyngeal dystrophy is a distinct autosomal dominant disease with onset in middle age, characterized by ptosis, limited eye movements, and trouble swallowing. Myotonic dystrophy, another autosomal dominant disorder, causes ptosis, ophthalmoparesis, cataract, and pigmentary retinopathy. Patients have muscle wasting, myotonia, frontal balding, and cardiac abnormalities.
Neurogenic Ptosis This results from a lesion affecting the innervation to either of the two muscles that open the eyelid: Müller’s muscle or the levator palpebrae superioris. Examination of the pupil helps distinguish between these two possibilities. In Horner’s syndrome, the eye with ptosis has a smaller pupil and the eye movements are full. In an oculomotor nerve palsy, the eye with the ptosis has a larger or a normal pupil. If the pupil is normal but there is limitation of adduction, elevation, and depression, a pupil-sparing oculomotor nerve palsy is likely (see next section). Rarely, a lesion affecting the small, central subnucleus of the oculomotor complex will cause bilateral ptosis with normal eye movements and pupils.
DOUBLE VISION (DIPLOPIA)
The first point to clarify is whether diplopia persists in either eye after the opposite eye is covered. If it does, the diagnosis is monocular diplopia. The cause is usually intrinsic to the eye and therefore has no dire implications for the patient. Corneal aberrations (e.g., keratoconus, pterygium), uncorrected refractive error, cataract, or foveal traction may give rise to monocular diplopia. Occasionally it is a symptom of malingering or psychiatric disease. Diplopia alleviated by covering one eye is binocular diplopia and is caused by disruption of ocular alignment. Inquiry should be made into the nature of the double vision (purely side-by-side versus partial vertical displacement of images), mode of onset, duration, intermittency, diurnal variation, and associated neurologic or systemic symptoms. If the patient has diplopia while being examined, motility testing should reveal a deficiency corresponding to the patient’s symptoms. However, subtle limitation of ocular excursions is often difficult to detect. For example, a patient with a slight left abducens nerve paresis may appear to have full eye movements despite a complaint of horizontal diplopia upon looking to the left. In this situation, the cover test provides a more sensitive method for demonstrating the ocular misalignment. It should be conducted in primary gaze and then with the head turned and tilted in each direction. In the above example, a cover test with the head turned to the right will maximize the fixation shift evoked by the cover test.
Occasionally, a cover test performed in an asymptomatic patient during a routine examination will reveal an ocular deviation. If the eye movements are full and the ocular misalignment is equal in all directions of gaze (concomitant deviation), the diagnosis is strabismus. In this condition, which affects about 1% of the population, fusion is disrupted in infancy or early childhood. To avoid diplopia, vision is suppressed from the nonfixating eye. In some children, this leads to impaired vision (amblyopia, or “lazy” eye) in the deviated eye.
Binocular diplopia results from a wide range of processes: infectious, neoplastic, metabolic, degenerative, inflammatory, and vascular. One must decide whether the diplopia is neurogenic in origin or is due to restriction of globe rotation by local disease in the orbit. Orbital pseudotumor, myositis, infection, tumor, thyroid disease, and muscle entrapment (e.g., from a blowout fracture) cause restrictive diplopia. The diagnosis of restriction is usually made by recognizing other associated signs and symptoms of local orbital disease. Omission of high-resolution orbital imaging is a common mistake in the evaluation of diplopia.
Myasthenia Gravis (See also Chap. 461) This is a major cause of diplopia. The diplopia is often intermittent, variable, and not confined to any single ocular motor nerve distribution. The pupils are always normal. Fluctuating ptosis may be present. Many patients have a purely ocular form of the disease, with no evidence of systemic muscular weakness. The diagnosis can be confirmed by an IV edrophonium injection, which produces a transient reversal of eyelid or eye muscle weakness. Blood tests for antibodies against the acetylcholine receptor or the MuSK protein can establish the diagnosis but are frequently negative in the purely ocular form of myasthenia gravis. Botulism from food or wound poisoning can mimic ocular myasthenia.
After restrictive orbital disease and myasthenia gravis are excluded, a lesion of a cranial nerve supplying innervation to the extraocular muscles is the most likely cause of binocular diplopia.
Oculomotor Nerve The third cranial nerve innervates the medial, inferior, and superior recti; inferior oblique; levator palpebrae superioris; and the iris sphincter. Total palsy of the oculomotor nerve causes ptosis, a dilated pupil, and leaves the eye “down and out” because of the unopposed action of the lateral rectus and superior oblique. This combination of findings is obvious. More challenging is the diagnosis of early or partial oculomotor nerve palsy. In this setting any combination of ptosis, pupil dilation, and weakness of the eye muscles supplied by the oculomotor nerve may be encountered. Frequent serial examinations during the evolving phase of the palsy help ensure that the diagnosis is not missed. The advent of an oculomotor nerve palsy with a pupil involvement, especially when accompanied by pain, suggests a compressive lesion, such as a tumor or circle of Willis aneurysm. Neuroimaging should be obtained, along with a CT or MR angiogram. Occasionally, a catheter arteriogram must be done to exclude an aneurysm.
A lesion of the oculomotor nucleus in the rostral midbrain produces signs that differ from those caused by a lesion of the nerve itself. There is bilateral ptosis because the levator muscle is innervated by a single central subnucleus. There is also weakness of the contralateral superior rectus, because it is supplied by the oculomotor nucleus on the other side. Occasionally both superior recti are weak. Isolated nuclear oculomotor palsy is rare. Usually neurologic examination reveals additional signs that suggest brainstem damage from infarction, hemorrhage, tumor, or infection.
Injury to structures surrounding fascicles of the oculomotor nerve descending through the midbrain has given rise to a number of classic eponymic designations. In Nothnagel’s syndrome, injury to the superior cerebellar peduncle causes ipsilateral oculomotor palsy and contralateral cerebellar ataxia. In Benedikt’s syndrome, injury to the red nucleus results in ipsilateral oculomotor palsy and contralateral tremor, chorea, and athetosis. Claude’s syndrome incorporates features of both of these syndromes, by injury to both the red nucleus and the superior cerebellar peduncle. Finally, in Weber’s syndrome, injury to the cerebral peduncle causes ipsilateral oculomotor palsy with contralateral hemiparesis.
In the subarachnoid space the oculomotor nerve is vulnerable to aneurysm, meningitis, tumor, infarction, and compression. In cerebral herniation, the nerve becomes trapped between the edge of the tentorium and the uncus of the temporal lobe. Oculomotor palsy also can result from midbrain torsion and hemorrhages during herniation. In the cavernous sinus, oculomotor palsy arises from carotid aneurysm, carotid cavernous fistula, cavernous sinus thrombosis, tumor (pituitary adenoma, meningioma, metastasis), herpes zoster infection, and the Tolosa-Hunt syndrome.
The etiology of an isolated, pupil-sparing oculomotor palsy often remains an enigma even after neuroimaging and extensive laboratory testing. Most cases are thought to result from microvascular infarction of the nerve somewhere along its course from the brainstem to the orbit. Usually the patient complains of pain. Diabetes, hypertension, and vascular disease are major risk factors. Spontaneous recovery over a period of months is the rule. If this fails to occur or if new findings develop, the diagnosis of microvascular oculomotor nerve palsy should be reconsidered. Aberrant regeneration is common when the oculomotor nerve is injured by trauma or compression (tumor, aneurysm). Miswiring of sprouting fibers to the levator muscle and the rectus muscles results in elevation of the eyelid upon downgaze or adduction. The pupil also constricts upon attempted adduction, elevation, or depression of the globe. Aberrant regeneration is not seen after oculomotor palsy from microvascular infarct and hence vitiates that diagnosis.
Trochlear Nerve The fourth cranial nerve originates in the midbrain, just caudal to the oculomotor nerve complex. Fibers exit the brainstem dorsally and cross to innervate the contralateral superior oblique. The principal actions of this muscle are to depress and intort the globe. A palsy therefore results in hypertropia and excyclotorsion. The cyclotorsion seldom is noticed by patients. Instead, they complain of vertical diplopia, especially upon reading or looking down. The vertical diplopia also is exacerbated by tilting the head toward the side with the muscle palsy and alleviated by tilting it away. This “head tilt test” is a cardinal diagnostic feature.
Isolated trochlear nerve palsy results from all the causes listed above for the oculomotor nerve except aneurysm. The trochlear nerve is particularly apt to suffer injury after closed head trauma. The free edge of the tentorium is thought to impinge on the nerve during a concussive blow. Most isolated trochlear nerve palsies are idiopathic and hence are diagnosed by exclusion as “microvascular.” Spontaneous improvement occurs over a period of months in most patients. A base-down prism (conveniently applied to the patient’s glasses as a stick-on Fresnel lens) may serve as a temporary measure to alleviate diplopia. If the palsy does not resolve, the eyes can be realigned by weakening the inferior oblique muscle.
Abducens Nerve The sixth cranial nerve innervates the lateral rectus muscle. A palsy produces horizontal diplopia, worse on gaze to the side of the lesion. A nuclear lesion has different consequences, because the abducens nucleus contains interneurons that project via the medial longitudinal fasciculus to the medial rectus subnucleus of the contralateral oculomotor complex. Therefore, an abducens nuclear lesion produces a complete lateral gaze palsy from weakness of both the ipsilateral lateral rectus and the contralateral medial rectus. Foville’s syndrome after dorsal pontine injury includes lateral gaze palsy, ipsilateral facial palsy, and contralateral hemiparesis incurred by damage to descending corticospinal fibers. Millard-Gubler syndrome from ventral pontine injury is similar except for the eye findings. There is lateral rectus weakness only, instead of gaze palsy, because the abducens fascicle is injured rather than the nucleus. Infarct, tumor, hemorrhage, vascular malformation, and multiple sclerosis are the most common etiologies of brainstem abducens palsy.
After leaving the ventral pons, the abducens nerve runs forward along the clivus to pierce the dura at the petrous apex, where it enters the cavernous sinus. Along its subarachnoid course it is susceptible to meningitis, tumor (meningioma, chordoma, carcinomatous meningitis), subarachnoid hemorrhage, trauma, and compression by aneurysm or dolichoectatic vessels. At the petrous apex, mastoiditis can produce deafness, pain, and ipsilateral abducens palsy (Gradenigo’s syndrome). In the cavernous sinus, the nerve can be affected by carotid aneurysm, carotid cavernous fistula, tumor (pituitary adenoma, meningioma, nasopharyngeal carcinoma), herpes infection, and Tolosa-Hunt syndrome.
Unilateral or bilateral abducens palsy is a classic sign of raised intracranial pressure. The diagnosis can be confirmed if papilledema is observed on fundus examination. The mechanism is still debated but probably is related to rostral-caudal displacement of the brainstem. The same phenomenon accounts for abducens palsy from Chiari malformation or low intracranial pressure (e.g., after lumbar puncture, spinal anesthesia, or spontaneous dural cerebrospinal fluid leak).
Treatment of abducens palsy is aimed at prompt correction of the underlying cause. However, the cause remains obscure in many instances despite diligent evaluation. As was mentioned above for isolated trochlear or oculomotor palsy, most cases are assumed to represent microvascular infarcts because they often occur in the setting of diabetes or other vascular risk factors. Some cases may develop as a postinfectious mononeuritis (e.g., after a viral flu). Patching one eye, occluding one eyeglass lens with tape, or applying a temporary prism will provide relief of diplopia until the palsy resolves. If recovery is incomplete, eye muscle surgery nearly always can realign the eyes, at least in primary position. A patient with an abducens palsy that fails to improve should be reevaluated for an occult etiology (e.g., chordoma, carcinomatous meningitis, carotid cavernous fistula, myasthenia gravis). Skull base tumors are easily missed even on contrast-enhanced neuroimaging studies.
Multiple Ocular Motor Nerve Palsies These should not be attributed to spontaneous microvascular events affecting more than one cranial nerve at a time. This remarkable coincidence does occur, especially in diabetic patients, but the diagnosis is made only in retrospect after all other diagnostic alternatives have been exhausted. Neuroimaging should focus on the cavernous sinus, superior orbital fissure, and orbital apex, where all three ocular motor nerves are in close proximity. In a diabetic or immunocompromised host, fungal infection (Aspergillus, Mucorales, Cryptococcus) is a common cause of multiple nerve palsies. In a patient with systemic malignancy, carcinomatous meningitis is a likely diagnosis. Cytologic examination may be negative despite repeated sampling of the cerebrospinal fluid. The cancer-associated Lambert-Eaton myasthenic syndrome also can produce ophthalmoplegia. Giant cell (temporal) arteritis occasionally manifests as diplopia from ischemic palsies of extraocular muscles. Fisher’s syndrome, an ocular variant of Guillain-Barré, produces ophthalmoplegia with areflexia and ataxia. Often the ataxia is mild, and the reflexes are normal. Antiganglioside antibodies (GQ1b) can be detected in about 50% of cases.
Supranuclear Disorders of Gaze These are often mistaken for multiple ocular motor nerve palsies. For example, Wernicke’s encephalopathy can produce nystagmus and a partial deficit of horizontal and vertical gaze that mimics a combined abducens and oculomotor nerve palsy. The disorder occurs in malnourished or alcoholic patients and can be reversed by thiamine. Infarct, hemorrhage, tumor, multiple sclerosis, encephalitis, vasculitis, and Whipple’s disease are other important causes of supranuclear gaze palsy. Disorders of vertical gaze, especially downward saccades, are an early feature of progressive supranuclear palsy. Smooth pursuit is affected later in the course of the disease. Parkinson’s disease, Huntington’s disease, and olivopontocerebellar degeneration also can affect vertical gaze.
The frontal eye field of the cerebral cortex is involved in generation of saccades to the contralateral side. After hemispheric stroke, the eyes usually deviate toward the lesioned side because of the unopposed action of the frontal eye field in the normal hemisphere. With time, this deficit resolves. Seizures generally have the opposite effect: the eyes deviate conjugately away from the irritative focus. Parietal lesions disrupt smooth pursuit of targets moving toward the side of the lesion. Bilateral parietal lesions produce Bálint’s syndrome, which is characterized by impaired eye-hand coordination (optic ataxia), difficulty initiating voluntary eye movements (ocular apraxia), and visuospatial disorientation (simultanagnosia).
Horizontal Gaze Descending cortical inputs mediating horizontal gaze ultimately converge at the level of the pons. Neurons in the paramedian pontine reticular formation are responsible for controlling conjugate gaze toward the same side. They project directly to the ipsilateral abducens nucleus. A lesion of either the paramedian pontine reticular formation or the abducens nucleus causes an ipsilateral conjugate gaze palsy. Lesions at either locus produce nearly identical clinical syndromes, with the following exception: vestibular stimulation (oculocephalic maneuver or caloric irrigation) will succeed in driving the eyes conjugately to the side in a patient with a lesion of the paramedian pontine reticular formation but not in a patient with a lesion of the abducens nucleus.
INTERNUCLEAR OPHTHALMOPLEGIA This results from damage to the medial longitudinal fasciculus ascending from the abducens nucleus in the pons to the oculomotor nucleus in the midbrain (hence, “internuclear”). Damage to fibers carrying the conjugate signal from abducens interneurons to the contralateral medial rectus motoneurons results in a failure of adduction on attempted lateral gaze. For example, a patient with a left internuclear ophthalmoplegia (INO) will have slowed or absent adducting movements of the left eye (Fig. 39-20). A patient with bilateral injury to the medial longitudinal fasciculus will have bilateral INO. Multiple sclerosis is the most common cause, although tumor, stroke, trauma, or any brainstem process may be responsible. One-and-a-half syndrome is due to a combined lesion of the medial longitudinal fasciculus and the abducens nucleus on the same side. The patient’s only horizontal eye movement is abduction of the eye on the other side.
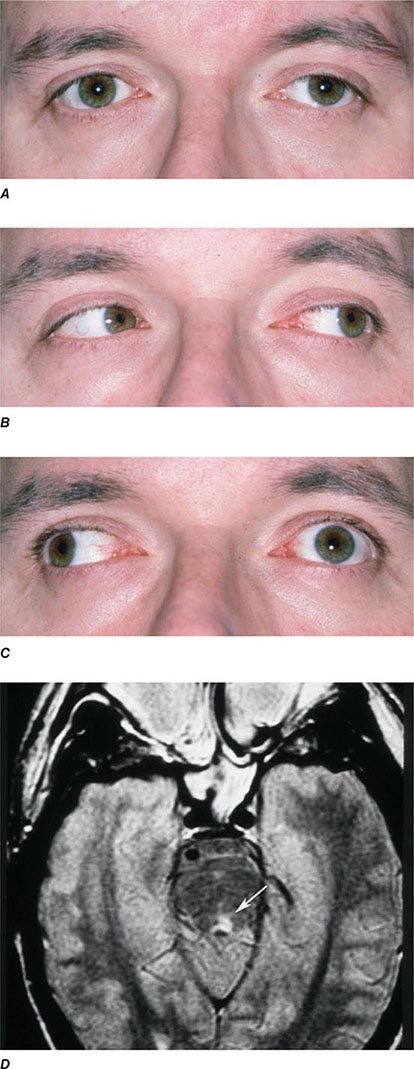
FIGURE 39-20 Left internuclear ophthalmoplegia (INO). A. In primary position of gaze, the eyes appear normal. B. Horizontal gaze to the left is intact. C. On attempted horizontal gaze to the right, the left eye fails to adduct. In mildly affected patients, the eye may adduct partially or more slowly than normal. Nystagmus is usually present in the abducted eye. D. T2-weighted axial magnetic resonance image through the pons showing a demyelinating plaque in the left medial longitudinal fasciculus (arrow).
Vertical Gaze This is controlled at the level of the midbrain. The neuronal circuits affected in disorders of vertical gaze are not fully elucidated, but lesions of the rostral interstitial nucleus of the medial longitudinal fasciculus and the interstitial nucleus of Cajal cause supranuclear paresis of upgaze, downgaze, or all vertical eye movements. Distal basilar artery ischemia is the most common etiology. Skew deviation refers to a vertical misalignment of the eyes, usually constant in all positions of gaze. The finding has poor localizing value because skew deviation has been reported after lesions in widespread regions of the brainstem and cerebellum.
PARINAUD’S SYNDROME Also known as dorsal midbrain syndrome, this is a distinct supranuclear vertical gaze disorder caused by damage to the posterior commissure. It is a classic sign of hydrocephalus from aqueductal stenosis. Pineal region or midbrain tumors, cysticercosis, and stroke also cause Parinaud’s syndrome. Features include loss of upgaze (and sometimes downgaze), convergence-retraction nystagmus on attempted upgaze, downward ocular deviation (“setting sun” sign), lid retraction (Collier’s sign), skew deviation, pseudoabducens palsy, and light-near dissociation of the pupils.
Nystagmus This is a rhythmic oscillation of the eyes, occurring physiologically from vestibular and optokinetic stimulation or pathologically in a wide variety of diseases (Chap. 28). Abnormalities of the eyes or optic nerves, present at birth or acquired in childhood, can produce a complex, searching nystagmus with irregular pendular (sinusoidal) and jerk features. Examples are albinism, Leber’s congenital amaurosis, and bilateral cataract. This nystagmus is commonly referred to as congenital sensory nystagmus. This is a poor term because even in children with congenital lesions, the nystagmus does not appear until weeks after birth. Congenital motor nystagmus, which looks similar to congenital sensory nystagmus, develops in the absence of any abnormality of the sensory visual system. Visual acuity also is reduced in congenital motor nystagmus, probably by the nystagmus itself, but seldom below a level of 20/200.
JERK NYSTAGMUS This is characterized by a slow drift off the target, followed by a fast corrective saccade. By convention, the nystagmus is named after the quick phase. Jerk nystagmus can be downbeat, upbeat, horizontal (left or right), and torsional. The pattern of nystagmus may vary with gaze position. Some patients will be oblivious to their nystagmus. Others will complain of blurred vision or a subjective to-and-fro movement of the environment (oscillopsia) corresponding to the nystagmus. Fine nystagmus may be difficult to see on gross examination of the eyes. Observation of nystagmoid movements of the optic disc on ophthalmoscopy is a sensitive way to detect subtle nystagmus.
GAZE-EVOKED NYSTAGMUS This is the most common form of jerk nystagmus. When the eyes are held eccentrically in the orbits, they have a natural tendency to drift back to primary position. The subject compensates by making a corrective saccade to maintain the deviated eye position. Many normal patients have mild gaze-evoked nystagmus. Exaggerated gaze-evoked nystagmus can be induced by drugs (sedatives, anticonvulsants, alcohol); muscle paresis; myasthenia gravis; demyelinating disease; and cerebellopontine angle, brainstem, and cerebellar lesions.
VESTIBULAR NYSTAGMUS Vestibular nystagmus results from dysfunction of the labyrinth (Ménière’s disease), vestibular nerve, or vestibular nucleus in the brainstem. Peripheral vestibular nystagmus often occurs in discrete attacks, with symptoms of nausea and vertigo. There may be associated tinnitus and hearing loss. Sudden shifts in head position may provoke or exacerbate symptoms.
DOWNBEAT NYSTAGMUS Downbeat nystagmus results from lesions near the craniocervical junction (Chiari malformation, basilar invagination). It also has been reported in brainstem or cerebellar stroke, lithium or anticonvulsant intoxication, alcoholism, and multiple sclerosis. Upbeat nystagmus is associated with damage to the pontine tegmentum from stroke, demyelination, or tumor.
Opsoclonus This rare, dramatic disorder of eye movements consists of bursts of consecutive saccades (saccadomania). When the saccades are confined to the horizontal plane, the term ocular flutter is preferred. It can result from viral encephalitis, trauma, or a paraneoplastic effect of neuroblastoma, breast carcinoma, and other malignancies. It has also been reported as a benign, transient phenomenon in otherwise healthy patients.
40e | Use of the Hand-Held Ophthalmoscope |
Examination of the living human retina provides a unique opportunity for the direct study of nervous, vascular, and connective tissues. Many systemic disorders have retinal manifestations that are valuable for screening, diagnosis, and management of these conditions. Furthermore, retinal involvement in systemic disorders, such as diabetes mellitus, is a major cause of morbidity. Early recognition by ophthalmoscopic screening is a key factor in effective treatment. Ophthalmoscopy has the potential to be one of the most “high-yield” elements of the physical examination. Effective ophthalmoscopy requires a basic understanding of ocular structures and ophthalmoscopic techniques and recognition of abnormal findings.
OVERVIEW OF OCULAR STRUCTURES
The eye consists of a shell (cornea and sclera), lens, iris diaphragm, ciliary body, choroid, and retina. The anterior chamber is the space between the cornea and the lens, and it is filled with aqueous humor. The space between the posterior aspect of the lens and the retina is filled by vitreous gel. The choroid and the retina cover the posterior two-thirds of the sclera internally. The cornea and the lens form the focusing system of the eye, while the retina functions as the photoreceptor system, translating light to neuronal signals that are in turn transmitted to the brain via the optic nerve and visual pathways. The choroid is a layer of highly vascularized tissue that nourishes the retina and is located between the sclera and the retina. The retinal pigment epithelium (RPE) layer is a monolayer of pigmented cells that are adherent to the overlying retinal photoreceptor cells. RPE plays a major role in retinal photoreceptor metabolism.
NORMAL FUNDUS
The important areas that are visible by ophthalmoscopy include the macula, optic disc, retinal blood vessels, and retinal periphery (Fig. 40e-1).
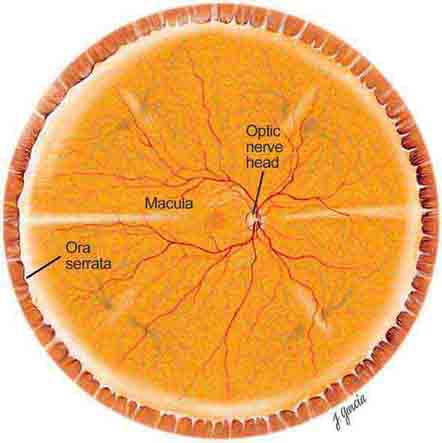
FIGURE 40e-1 Diagram showing the landmarks of the normal fundus. The macula is bounded by the superior and inferior vascular arcades and extends for 5 disc diameters (DD) temporal to the optic disc (optic nerve head). The central part of the macula (fovea) is located 2.5 DD temporal to the optic disc. The peripheral fundus is arbitrarily defined as the area extending anteriorly from the opening of the vortex veins to the ora serrata (the juncture between the retina and ciliary body). (Drawing courtesy of Juan R. Garcia. Used with permission from Johns Hopkins University.)
THE MACULA
The macula is the central part of the retina and is responsible for detailed vision (acuity) and perception of color. The macula is defined clinically as the area of the retina centered on the posterior pole of the fundus, measuring about 5 disc diameters (DD) (7–8 mm) and bordered by the optic disc nasally and the temporal vascular arcades superiorly and inferiorly. Temporally, the macula extends for about 2.5 DD from its center. The fovea, in the central part of the macula, corresponds to the site of sharpest visual acuity. It is approximately 1 DD in size and appears darker in color than the surrounding area. The center of the fovea, the foveola, has a depressed pit-like configuration measuring about 350 μm.
THE OPTIC DISC
The optic disc measures about 1.5 mm and is located about 4 mm (2.5 DD) nasal to the fovea. It contains the central retinal artery and vein as they branch, a central excavation (cup), and a peripheral neural rim. Normally, the cup-to-disc ratio is less than 0.6. The cup is located temporal to the entry of the disc vessels. The normal optic disc is yellow/pink in color. It has clear and well-defined margins and is in the same plane as the retina (Fig. 40e-2). Pathologic findings include pallor (atrophy), swelling, and enlarged cupping.
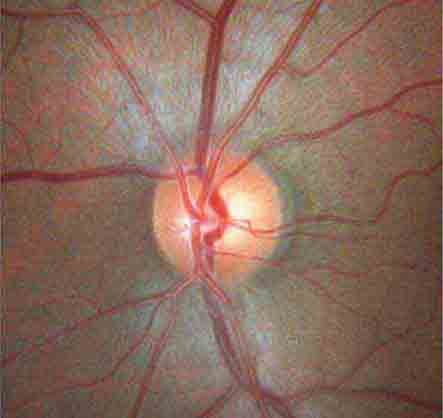
FIGURE 40e-2 Photograph of a normal left optic disc illustrating branching of the central retinal vein and artery, a physiologic cup, surface capillaries, and distinct margin. The cup is located temporal to the entry of the disc vessels. (From H Tabandeh, MF Goldberg: Retina in Systemic Disease: A Color Manual of Ophthalmoscopy. New York, Thieme, 2009.)
THE EQUATOR AND PERIPHERAL RETINA
The equator of the fundus is clinically defined as the area that includes the internal opening of the vortex veins. The peripheral retina extends from the equator anteriorly to the ora serrata.
OPHTHALMOSCOPY
There are a number of ways to visualize the retina, including direct ophthalmoscopy, binocular indirect ophthalmoscopy, and slit-lamp biomicroscopy. Most nonophthalmologists prefer direct ophthalmoscopy, performed with a hand-held ophthalmoscope, because the technique is simple to master and the device is very portable. Ophthalmologists often use slit-lamp biomicroscopy and indirect ophthalmoscopy to obtain a more extensive view of the fundus.
DIRECT OPHTHALMOSCOPE
Direct ophthalmoscopes are simple hand-held devices that include a small light source for illumination, a viewing aperture through which the examiner looks at the retina, and a lens dial used for correction of the examiner’s and the patient’s refractive errors. A more recent design, the PanOptic ophthalmoscope, provides a wider field of view.
How to Use a Direct Ophthalmoscope Good alignment is the key. The goal is to align the examiner’s eye with the viewing aperture of the ophthalmoscope, the patient’s pupil, and the area of interest on the retina. Both the patient and the examiner should be in a comfortable position (sitting or lying for the patient, sitting or standing for the examiner). Dilating the pupil and dimming the room lights make the examination easier. Steps for performing direct ophthalmoscopy are summarized in Table 40e-1.
GUIDELINES FOR PERFORMING DIRECT OPHTHALMOSCOPY |
PANOPTIC OPHTHALMOSCOPE
The PanOptic ophthalmoscope is a type of direct ophthalmoscope that is designed to provide a wider view of the fundus and has slightly more magnification than the standard direct ophthalmoscope. Steps for using the PanOptic Ophthalmoscope are summarized in Table 40e-2.
HOW TO USE A PANOPTIC OPHTHALMOSCOPE |
RETINAL SIGNS ASSOCIATED WITH SYSTEMIC DISEASES
AGE-RELATED CHANGES
Common age-related changes include diminished foveal light reflex, drusen (small yellow subretinal deposits), mild RPE atrophy, and pigment clumping.
RETINAL HEMORRHAGES
Retinal hemorrhages may take various shapes and sizes depending on their location within the retina (Figs. 40e-3 and 40e-4). Flame-shaped hemorrhages are located at the level of the superficial nerve fiber layer and represent bleeding from the inner capillary network of the retina. A white-centered hemorrhage is a superficial flame-shaped hemorrhage with an area of central whitening, often representing edema, focal necrosis, or cellular infiltration. Causes of white-centered hemorrhage include bacterial endocarditis and septicemia (Roth spots), lymphoproliferative disorders, diabetes mellitus, hypertension, anemia, and collagen vascular disorders. Dot hemorrhages are small, round, superficial hemorrhages that also originate from the superficial capillary network of the retina. They resemble microaneurysms. Blot hemorrhages are slightly larger in size, dark, and intraretinal. They represent bleeding from the deep capillary network of the retina. Subhyaloid hemorrhages are variable in shape and size and tend to be larger than other types of hemorrhages. They often have a fluid level (“boat-shaped” hemorrhage) and are located within the space between the vitreous and the retina. Subretinal hemorrhages are located deep (external) to the retina. The retinal vessels can be seen crossing over (internal to) such hemorrhages. Subretinal hemorrhages are variable in size and most commonly are caused by choroidal neovascularization (e.g., wet macular degeneration).
FIGURE 40e-3 Superficial flame-shaped hemorrhages, dot hemorrhages, and microaneurysms in a patient with nonproliferative diabetic retinopathy.
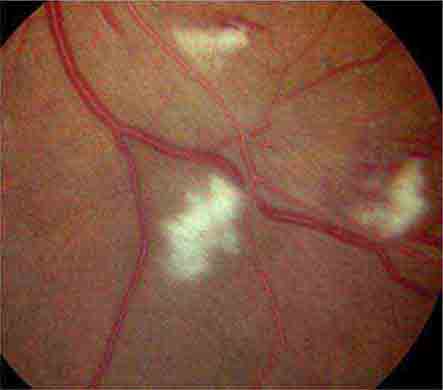
FIGURE 40e-4 Deep and superficial retinal hemorrhages in a patient with chronic leukemia.
Conditions associated with retinal hemorrhages include diseases causing retinal microvasculopathy (Table 40e-3), retinitis, retinal macroaneurysm, papilledema, subarachnoid hemorrhage (Terson’s syndrome), Valsalva retinopathy, trauma (ocular injury, head injury, compression injuries of chest and abdomen, shaken baby syndrome, strangulation), macular degeneration, and posterior vitreous detachment. Hyperviscosity states may produce dot and blot hemorrhages, dilated veins (“string of sausages” appearance), optic disc edema, and exudates; similar changes can occur with adaptation to high altitude in mountain climbers.
DISEASES ASSOCIATED WITH RETINAL MICROVASCULOPATHY |
MICROANEURYSMS
Microaneurysms are outpouchings of the retinal capillaries, appearing as red dots (similar to dot hemorrhages) and measuring 15–50 μm. Microaneurysms have increased permeability and may bleed or leak, resulting in localized retinal hemorrhage or edema. A microaneurysm ultimately thromboses and disappears within 3–6 months. Microaneurysms may occur in any condition that causes retinal microvasculopathy (Table 40e-3).
HARD EXUDATES
Hard exudates are well-circumscribed, shiny, yellow deposits located within the retina. They arise at the margins of areas of retinal edema and indicate increased capillary permeability. Hard exudates contain lipoproteins and lipid-laden macrophages. They may clear spontaneously or following laser photocoagulation, often within 6 months. Hard exudates may occur in isolation or may be scattered throughout the fundus. They may occur in a circular (circinate) pattern centered around an area of leaking microaneurysms. A macular star consists of a radiating, star-shaped pattern of hard exudates that is characteristically seen in severe systemic hypertension and in neuroretinitis associated with cat-scratch disease. Conditions associated with hard exudates include those causing retinal microvasculopathy (Table 40e–3), papilledema, neuroretinitis such as cat-scratch disease and Lyme disease, retinal vascular lesions (macroaneurysm, retinal capillary hemangioma, Coats’ disease), intraocular tumors, and wet age-related macular degeneration. Drusen may be mistaken for hard exudates on ophthalmoscopy. Unlike hard exudates, drusen are nonrefractile subretinal deposits with blurred margins. They are usually seen in association with age-related macular degeneration.
COTTON-WOOL SPOTS
Cotton-wool spots are yellow/white superficial retinal lesions with indistinct feathery borders measuring 0.25–1 DD in size (Fig. 40e-5). They represent areas of edema within the retinal nerve fiber layer due to focal ischemia. Cotton-wool spots usually resolve spontaneously within 3 months. If the underlying ischemic condition persists, new lesions can develop in different locations. Cotton-wool spots often occur in conjunction with retinal hemorrhages and microaneurysms and represent retinal microvasculopathy caused by a number of systemic conditions (Table 40e-3). They may occur in isolation in HIV retinopathy, systemic lupus erythematosus, anemia, bodily trauma, other systemic conditions (Purtscher’s/Purtscher’s-like retinopathy), and interferon therapy.
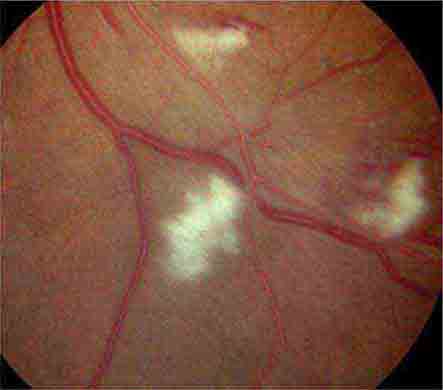
FIGURE 40e-5 Cotton-wool spots, yellow-white superficial lesions with characteristic feathery borders, in a patient with hypertensive retinopathy. (From H Tabandeh, MF Goldberg: Retina in Systemic Disease: A Color Manual of Ophthalmoscopy. New York, Thieme, 2009.)
RETINAL NEOVASCULARIZATION
Retinal neovascular complexes are irregular meshworks of fine blood vessels that grow in response to severe retinal ischemia or chronic inflammation (Fig. 40e-6). They may occur on or adjacent to the optic disc or elsewhere in the retina. Neovascular complexes are very fragile and have a high risk for hemorrhaging, often causing visual loss. Diseases associated with retinal neovascularization include conditions that cause severe retinal microvasculopathy, especially diabetic and sickle cell retinopathies (Table 40e-3), intraocular tumors, intraocular inflammation (sarcoidosis, chronic uveitis), and chronic retinal detachment.
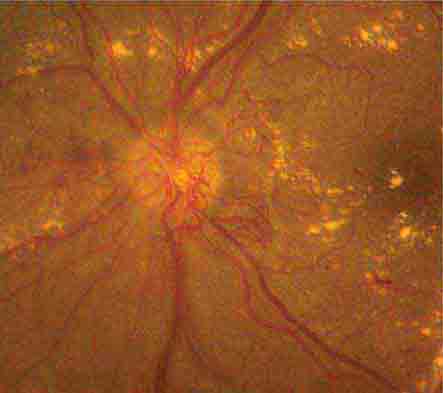
FIGURE 40e-6 Optic disc neovascularization in a patient with severe proliferative diabetic retinopathy. Multiple hard exudates are also present.
RETINAL EMBOLI
Common sources of retinal emboli include carotid artery atheromatous plaque, cardiac valve and septal abnormalities, cardiac arrhythmias, atrial myxoma, bacterial endocarditis, septicemia, fungemia, and intravenous drug abuse.
Platelet emboli are yellowish in appearance and conform to the shape of the blood vessel. They usually originate from an atheromatous plaque within the carotid artery and can cause transient loss of vision (amaurosis fugax). Cholesterol emboli, otherwise termed Hollenhorst plaques, are yellow crystalline deposits that are commonly found at the bifurcations of the retinal arteries and may be associated with amaurosis fugax. Calcific emboli have a pearly white appearance, are larger than the platelet and cholesterol emboli, and tend to lodge in the larger retinal arteries in or around the optic disc. Calcific emboli often result in retinal arteriolar occlusion. Septic emboli can cause white-centered retinal hemorrhages (Roth spots), retinal microabscesses, and endogenous endophthalmitis. Fat embolism and amniotic fluid embolism are characterized by multiple small vessel occlusions, typically causing cotton-wool spots and few hemorrhages (Purtscher’s-like retinopathy). Talc embolism occurs with intravenous drug abuse and is characterized by multiple refractile deposits within the small retinal vessels. Any severe form of retinal artery embolism may result in retinal ischemia and its sequelae, including retinal neovascularization.
CHERRY RED SPOT AT THE MACULA
Cherry red spot at the macula is the term used to describe the dark red appearance of the central foveal area in comparison to the surrounding macular region (Fig. 40e-7). This appearance is most commonly due to a relative loss of transparency of the parafoveal retina resulting from ischemic cloudy swelling or storage of macromolecules within the ganglion cell layer. Diseases associated with a cherry red spot at the macula include central retinal artery occlusion, sphingolipidoses, and mucolipidoses.
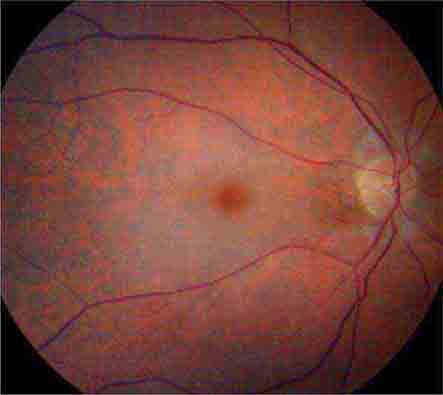
FIGURE 40e-7 Cherry red spot at the macula and cloudy swelling of the macula in a patient with central retinal artery occlusion due to embolus originating from a carotid artery atheromatous plaque.
RETINAL CRYSTAL DEPOSITION
Retinal crystals appear as fine, refractile, yellow-white deposits. Associated conditions include infantile cystinosis, primary hyperoxaluria, secondary oxalosis, Sjögren-Larson syndrome, intravenous drug abuse (talc retinopathy), and drugs such as tamoxifen, canthaxanthin, nitrofurantoin, methoxyflurane, and ethylene glycol. Crystals may also be seen in primary retinal diseases such as juxtafoveal telangiectasia, gyrate atrophy, and Bietti’s crystalline degeneration. Old microemboli may mimic retinal crystals.
RETINAL VASCULAR SHEATHING
Vascular sheathing appears as a yellow-white cuff surrounding a retinal artery or vein (Fig. 40e-8). Diseases associated with retinal vascular sheathing include sarcoidosis, tuberculosis, toxoplasmosis, syphilis, HIV, retinitis (cytomegalovirus, herpes zoster, and herpes simplex), Lyme disease, cat-scratch disease, multiple sclerosis, chronic leukemia, amyloidosis, Behçet’s disease, retinal vasculitis, retinal vascular occlusion, and chronic uveitis.
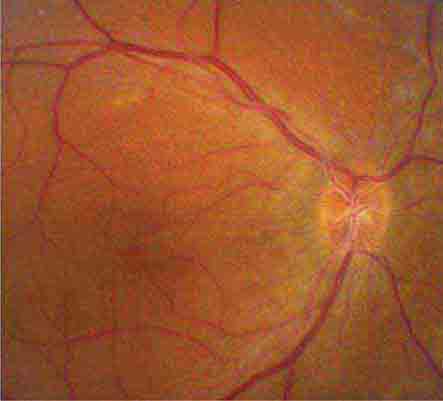
FIGURE 40e-8 Vascular sheathing over the optic disc in a patient with neurosarcoidosis.
RETINAL DETACHMENT
Retinal detachment is the separation of the retina from the underlying RPE. There are three main types: (1) serous/exudative, (2) tractional, and (3) rhegmatogenous retinal detachment.
In serous retinal detachment, the location of the subretinal fluid is position-dependent, characteristically gravitating to the lowermost part of the fundus (shifting fluid sign), and retinal breaks are absent. Diseases associated with serous/exudative retinal detachment include severe systemic hypertension, dural arteriovenous shunt, retinal vascular anomalies, hyperviscosity syndromes, papilledema, posterior uveitis, scleritis, orbital inflammation, and intraocular neoplasms such as choroidal melanoma, choroidal metastasis, lymphoma, and multiple myeloma.
Tractional retinal detachment is caused by internal traction on the retina in the absence of a retinal break. The retina in the area of detachment is immobile and concaved internally. Fibrovascular proliferation is a frequent associated finding. Conditions associated with tractional retinal detachment include vascular proliferative retinopathies such as severe proliferative diabetic retinopathy, branch retinal vein occlusion, sickle cell retinopathy, and retinopathy of prematurity. Ocular trauma, proliferative vitreoretinopathy, and intraocular inflammation are other causes of a tractional retinal detachment.
Rhegmatogenous retinal detachment is caused by the presence of a retinal break, allowing fluid from the vitreous cavity to gain access to the subretinal space. The surface of the retina is usually convex forward. Rhegmatogenous retinal detachment has a corrugated appearance, and undulates with eye movement. Causes of retinal breaks include posterior vitreous detachment, severe vitreoretinal traction, trauma, intraocular surgery, retinitis, and atrophic holes.
OPTIC DISC SWELLING
Optic disc swelling is abnormal elevation of the optic disc with blurring of its margins (Fig. 40e-9). The term “papilledema” is used to describe swelling of the optic disc secondary to elevation of intracranial pressure. In papilledema, the normal venous pulsation at the disc is characteristically absent. The differential diagnosis of optic disc swelling includes papilledema, anterior optic neuritis (papillitis), central retinal vein occlusion, anterior ischemic optic neuropathy, toxic optic neuropathy, hereditary optic neuropathy, neuroretinitis, diabetic papillopathy, hypertension (Fig. 40e-10), respiratory failure, carotid-cavernous fistula, optic disc nerve infiltration (glioma, lymphoma, leukemia, sarcoidosis, and granulomatous infections), ocular hypotony, chronic intraocular inflammation, optic disc drusen (pseudopapilledema), and high hypermetropia (pseudopapilledema).
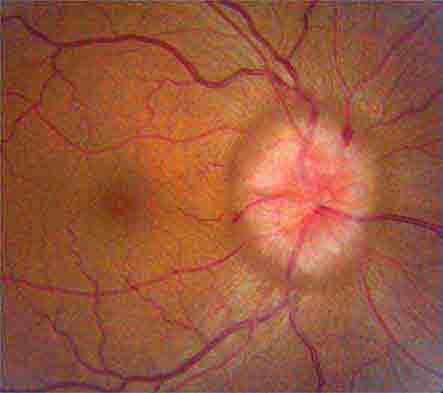
FIGURE 40e-9 Optic disc swelling in a patient with papilledema due to idiopathic intracranial hypertension. The optic disc is hyperemic, with indistinct margins. Superficial hemorrhages are present.
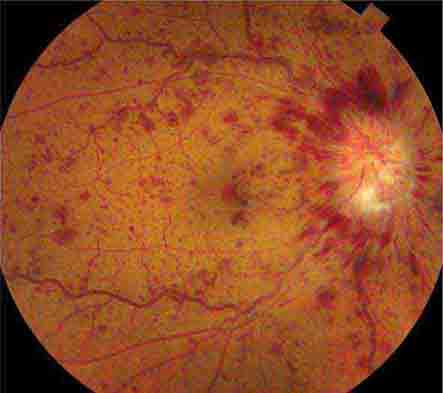
FIGURE 40e-10 Optic disc edema and retinal hemorrhages in a patient with malignant hypertension.
CHORIORETINAL MASS LESIONS
Choroidal mass lesions appear thickened and may or may not be associated with increased pigmentation. Pigmented mass lesions include choroidal nevus (usually flat), choroidal malignant melanoma (Fig. 40e-11), and melanocytoma. Nonpigmented lesions include amelanotic choroidal melanoma, choroidal metastasis, retinoblastoma, capillary hemangioma, granuloma (e.g., Toxocara canis), choroidal detachment, choroidal hemorrhage, and wet age-related macular degeneration. Other rare tumors that may be visible on ophthalmoscopy include osteoma, astrocytoma (e.g., tuberous sclerosis), neurilemmoma, and leiomyoma.
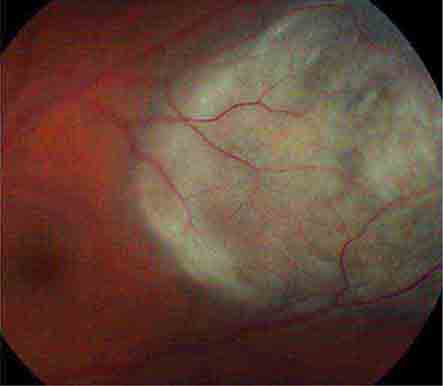
FIGURE 40e-11 Choroidal malignant melanoma. The lesion is highly elevated and pigmented, and has subretinal orange pigment deposits characteristic for malignant melanoma.
PIGMENTED LESIONS
The differential diagnosis of flat pigmented lesions of the fundus is summarized in Table 40e-4. The appearance of chorioretinal scarring from old Toxoplasma chorioretinitis is shown in Fig. 40e-12.
DIFFERENTIAL DIAGNOSIS OF FLAT PIGMENTED LESIONS OF THE FUNDUS |



