The pediatric surgical pathologist who is presented with a “soft tissue tumor” from a child may be confronted with a wide spectrum of pathology ranging from an enlarged lymph node, fibroinflammatory process in the superficial soft tissues, vascular malformation (VM) or a true neoplasm. Some soft tissue tumors (STTs) arise in the skin (infantile myofibroma) with involvement of the subcutis (infantile subcutaneous fibromatosis or lipofibromatosis), or at the level of the fascia and the deep soft tissues. Some of the soft-tissue sarcomas (STSs) in children may be organ based as in the case of embryonal rhabdomyosarcoma (ERMS) of the bladder or prostate gland, or congenital infantile fibrosarcoma (CIFS) of the small intestine, whereas others such as Ewing sarcoma-primitive neuroectodermal tumor (EWS-PNET), alveolar rhabdomyosarcoma (ARMS), and synovial sarcoma (SS), have the more familiar pattern of presenting in the deep soft tissues of the extremities. However, EWS-PNET is known to present in the kidney and SS can arise in the lung. Immunohistochemistry (IHC) and molecular diagnostic studies have facilitated the diagnostic evaluation of STSs in children of malignant and benign types (1). The fusion proteins from the various translocations among the STSs provide potential molecular targets for therapy (2).
One of the more common clinical diagnoses accompanying a pediatric surgical specimen from the superficial soft tissues is “ruleout soft-tissue tumor.” If this is the case, the neoplasm is benign in the majority of cases and more often than not is either a vascular anomaly of one type or another or a nonneoplastic process such as deep granuloma annulare. Of course, there is the dilemma in the case of some vascular tumors whether they are true neoplasms or malformations, which could have some bearing upon the surgical management. Vascular, neurogenic, fibrous myofibroblastic and myogenic tumors account for the majority (70% to 85%) of all STTs in children (3). Most of these tumors are benign (60% to 70% of cases) and have a predilection for the trunk, extremities, and head and neck regions in descending order of frequency; most of the benign STTs come to clinical attention at or before 10 years of age. Some of the more aggressive STSs, such as EWS-PNET, ARMS and SS, are usually diagnosed in the second decade of life; however, like all generalizations, there are exceptions and each one of these neoplasms has been diagnosed in children less than 5 years of life (4). Arguably one of the most malignant and treatment-resistant STSs of childhood is the malignant rhabdoid tumor (MRT) with its many organ-based primary sites (kidney, liver, and central nervous system), in addition to various nonvisceral soft-tissue locations such as the head and neck and mediastinum. Congenital STTs are mainly restricted to vascular anomalies, benign fibrous tumors of various types and teratomas arising in the sacrococcygeal soft tissues, retroperitoneum, and head and neck. Other less common STTs presenting at birth or in the first month of life are CIFS, myofibroma-myofibromatosis, granular cell tumor (GCT), primarily the congenital epulis of the oral cavity, ERMS, dermatofibrosarcoma protuberans (DFSP) and rarely ARMS. However, again, one should be prepared for the unanticipated when the clinical impression is a STT in a child.
The classification of STTs in children is accommodated for the most part by the World Health Organization (WHO) classification (5). Traditionally, STTs are classified on the basis of tissue differentiation and phenotype as determined by IHC. Molecular genetics has come to occupy an increasingly important role in the diagnosis of both benign and malignant STTs in children and adults for that matter (1,6) (Table 25-1). One of the first STS-specific, nonrandom chromosomal abnormalities, t(11;22)(q24;q12) translocation, was identified in EWS-PNET, the second most common STS of childhood (7). Emerging from this initial observation over the succeeding years has been the appreciation that there is a “family” of STSs with a predilection for children, adolescents, and young adults whose specific tumor types have nonrandom chromosomal translocations with the EWS gene on chromosome 22q as one of the fusion partners. This concept of families of tumors sharing a common fusion partner has evolved in the case of EWS, ALK, FUS and INI1 (8). These chromosomal perturbations can be detected by multiple techniques including (in approximate order of increasing sensitivity) conventional karyotyping, fluorescent in situ hybridization (FISH) on nuclear preparations from fresh or cultured tumor cells or formalin-fixed, paraffin-embedded tissue sections, DNA sequencing, or polymerase chain reaction (PCR).
TABLE 25-1 SOFT TISSUE NEOPLASMS IN CHILDREN AND THEIR RECURRING CYTOGENETIC ABNORMALITIES
The approach to the pathologic diagnosis of a STT from a child has the same starting point as one in an adult—a careful gross examination followed by the selection of representative tissue blocks based on the macroscopic features of the tumor (if one has digital photographic capability, a gross illustration can often substitute for a long narrative description; a picture is indeed worth a thousand words). The decision about the number of tissue blocks to submit is guided by the size of the specimen and the heterogeneity of gross features ranging from viable areas to those with a necrotic or hemorrhagic appearance. Many blocks may be required to identify any residual tumor in those cases with prior adjuvant therapy. If the specimen is submitted as a gross resection, the peripheral margins and any attached organs or bony structures should be identified and the margins tattooed with India ink or other dyes that will survive processing in order to evaluate the adequacy of the surgical margins. Margins of surgical resection are generally reported as “free of tumor” or not. Determining the distance between the tumor and the tumor-free margin by gross and microscopic examination is difficult in many cases. Some margins are limited by the constraints of the anatomy as it relates to neurovascular bundles or bony structures. The margins can be even more challenging in an infant or small child.
If the specimen is a small biopsy and submitted for intra-operative frozen section consultation, very little tissue may remain for permanent sections, let alone ancillary studies; thus, another tissue sample should be requested, if at all possible, as a contingency. Once the biopsy has been examined, it should be marked with an appropriate dye and placed in a small tea bag before tissue processing. If facilities are available and the tissue sample is judged to be more than adequate for histologic examination and molecular studies, tissue banking should be considered as well. Often times, the safest course of action is to snap-freeze a sugar cube-sized (approximately 1 cm3) piece of viable-appearing tissue and wait for the histologic examination to guide the selection of additional studies, most of which can be performed on a frozen specimen.
The world of STTs in children can be divided into three morphologic spheres: vascular tumors of varying morphology, spindle cell tumors, and small and not-so-small round cell tumors (Table 25-2). Various spindle cell tumors are listed in this tabulation and many of these are familiar to the pediatric pathologist. Some of the diagnoses are made with relative ease through one’s own experience with the appreciation of specific histologic details, which separate that particular round cell or spindle cell proliferation from all of the other similar appearing tumors. As one peruses Table 25-2, it becomes apparent that some of these STTs occur almost exclusively in the first two decades of life whereas others are seen more often in adolescence or young adulthood. Again, while the patient’s age should facilitate the crafting of a differential diagnosis, it should never be the sole cause for excluding a particular diagnosis.
Ancillary IHC studies have had a profound, impact upon the practice of surgical pathology over the past 25 years, but especially so in the diagnosis of soft tissue and hematopoietic neoplasms; the same can be said about the role of molecular genetics for these two phenotypic categories. In the case of malignancies in children, several of the more common neoplasms, soft tissue or otherwise, are morphologically similar from the perspective of their more or less uniform composition of small or large malignant round cells (Table 25-2). There are other accompanying features, which should be incorporated into the construction of the differential diagnosis without the need to utilize every commercially available antibody in one’s IHC laboratory (it does become necessary in some cases). However, it is acknowledged that there are those occasional cases in which successive waves of newly ordered IHC becomes necessary in order to arrive at a final diagnosis or the realization for the need of consultation. In the meantime, the titer of anxiety is on the rise for all concerned in the care of the patient. A difficult case is difficult for no other reason than that it is a difficult case as an existential reality or one simply does not appreciate the diagnosis. The most difficult diagnosis in the world to make is the one that we do not consider or it has yet to be described. Attention to the clinical aspects including clinical laboratory studies can be helpful in crafting the differential diagnosis and guiding the selection of stains. Table 25-3 summarizes the immunophenotype of malignant round cell tumors seen in children and should be familiar to most pathologists with some level of experience with the malignant round cell tumors of childhood. Each of these tumor types, less the ever diminishing category of “undifferentiated round cell sarcoma,” has one or more molecular aberrations, often with diagnostic and prognostic implications. One emerging category of malignant round cell neoplasms is the EWS-like sarcomas with their unique translocations, but with the morphology and immunophenotype of EWS-PNET.
TABLE 25-2 MORPHOLOGIC THEMES IN SSTS IN CHILDREN SOME DIFFERENTIAL DIAGNOSTIC CONSIDERATIONS
Morphology
Tumor Types
Round and not so “rounded” small cells
RMS (both ERMS and ARMS)
EWS-PNET
EWS-like sarcoma
Mononuclear histiocytic tumors (LCH, JXG, Rosai-Dorfman disease, DC tumor, giant cell tumor of tendon sheath, LCH and JXG
Hemangiomas of diverse subtypes and hemangioendothelioma
Vascular malformations
Lymphatic malformations
Cystic hygroma
There are other “round cell” neoplasms presenting in childhood, though chiefly in adults, which are not included in Table 25-2. Some of the round cell neoplasms have the added characterization of “epithelioid” whose cytomorphologic features are polygonal contours, a central nucleus, prominent nucleolus and abundant eosinophilic to clear cytoplasm. Alveolar soft part sarcoma (ASPS), perivascular epithelioid cell tumor, epithelioid vascular neoplasms, MRT and epithelioid sarcoma (ES) are among the more familiar tumor types in this category.
GRADING OF SOFT TISSUE SARCOMAS
STSs in adults are commonly graded pathologically on the basis of mitotic activity [mitoses per 10 or 50 high power fields (×400)], nuclear pleomorphism, and necrosis. Pleomorphic undifferentiated sarcoma (PUS) (formerly many of these sarcomas were interpreted as “malignant fibrous histiocytomas”), liposarcoma (LPS) and leiomyosarcoma are the three most common STSs in adults, which lend themselves to this traditional grading scheme (9). Pathologic grading of nonrhabdomyosarcomatous soft tissue tumors (NRMSTS) in the Children’s Oncology Group (COG) system (modified National Cancer Institute system) relies on a combination of morphologic type and histologic features: CIFS and myxoid LPS are grade 1, mesenchymal chondrosarcoma (MCS) and alveolar soft part sarcoma are grade 3, and other tumors are assigned either grade 2 or grade 3 depending on the mitotic activity and extent of necrosis (>5 mitoses per 10 high-powered fields (40× objective and/or >15% surface area necrosis indicate grade 3). Needless to say, differences in pathologic grading may arise between observers in a particular STS. A comparison between the COG and Fédération Nationale des Centre de Lutte Contre le Cancer (FNCLCC) systems of grading of STSs demonstrated that these systems were equally predictive for 5-year event free survivals except for the intermediate prognosis groups (9). Pathologic staging of STSs in children differs in several respects from sarcomas in adults and especially so in the case of RMSs in children where the primary site has a significant impact upon the stage and thus the prognosis as in the case of one unfavorable site, the perianal-perineal location.
TABLE 25-3 MALIGNANT ROUND CELL TUMORS AND THEIR IMMUNOPHENOTYPE
Some important developments in regard to STTs in children in the recent past include the reclassification of nodular fasciitis (NF) as a transient fibrous neoplasm on the basis of its molecular genetics, the stratification of RMSs on the basis of the presence or absence of PAX3 or PAX7 fusion partners with FOXO1 (FKHR) and the recognition of EWS-like round cell sarcomas with unique translocations other than an EWS fusion transcript (10,11,12).
VASCULAR TUMORS
Vascular anomalies, including both neoplasms and malformations, are the most common STTs in children, account for 20% to 30% of all cases and are among the most frequently recognized tumors at or shortly after birth and as many as one-third of cases are diagnosed in the first year of life (3,13,14,15). Cutaneous and even deep organ vascular anomalies are seen with multifocal sites of involvement in infants.
Traditionally, vasoformative lesions (or tumors) have been divided morphologically on the basis of their resemblance to blood vessels or lymphatics. A pathologic distinction is also made between a vascular neoplasm and malformation, which has been incorporated into the general classification of “vascular anomalies” (16). The International Society for the study of Vascular Anomalies (ISSVA) classification is more widely utilized by clinicians whereas the WHO classification of vascular tumors is more familiar to most general pathologists but the pediatric pathologist is generally expected to be conversant with the ISSVA classification and its application to vascular lesions in children (5) (Table 25-4). If one turns to the standard dermatopathology reference, the number of individual vascular lesions expands to another level (17).
Hemangioma with any number of qualifying prefixes is the most common pathologic diagnosis of STTs seen in the first two decades of life (3,13). The most common site is the skin where 25% or more of all vascular anomalies are encountered. There is a preference for the head and neck region in the case of skin and soft tissue involvement. Other sites include the deep soft tissues, bone, orbit, parotid gland, skeletal muscle, and upper air passages including the nasal cavity and larynx. The overwhelming majority (70% to 90%) of hemangiomas in children are initially recognized at or before 6 months of age (18). It is difficult in reviewing studies from the past to determine the proportion of infantile hemangiomas relative to VMs, also considered “hemangiomas” in many cases before the current conceptualization of the pathogenesis of vascular anomalies. However, one study noted that among 932 children with vascular anomalies, two-thirds were hemangiomas and the remainder were VMs (19).
TABLE 25-4 CLASSIFICATION OF VASCULAR ANOMALIES
ISSVA
WHO, 2013
Tumors
Hemangioma
Hemangiomas (benign)
Infantile – (GLUT-1+)
Synovial
Congenital (GLUT-1−)
Venous
Tufted angioma (D2-40+)
Arteriovenous
Epithelioid
Intramuscular
Spindle cell (D2-40+)
Capillary
Lobular capillary (pyogenic granuloma)
HE
Kaposiform (D2-40+)
Epithelioid
Papillary intralymphatic (Dabska tumor)
Angiomatosis
Retiform
Lymphangioma
Spindle cell
Intermediate, locally aggressive
Epithelioid
Kaposiform HEA
Angiosarcoma
Retiform HEA
Intermediate (rarely metastasizing)
Papillary intralymphatic angioendothelioma
Composite HEA
KS
Malignant
Epithelioid HEA
Angiosarcoma
Malformations
Simple (slow flow)
Arteriovenous malformations/hemangiomas
Capillary (portwine, angiokeratoma)
Lymphatic (lymphangioma)
Venous (cavernous hemangiomas)
Simple (fast flow)
Arterial (arteriovenous hemangiomas)
Combined
AVM
Capillary—venous
Capillary—lymphatic venous
Lymphatic—venous
Capillary AVM
INFANTILE AND CONGENITAL HEMANGIOMAS
Infantile hemangioma (IH) is the most common vascular anomaly in children, excluding lobular capillary hemangioma-pyogenic granuloma (LCH-PG) from the current discussion (19). A prospective study of 578 pregnant women and their 594 infants revealed that 29 (4.5%) infants had IHs (20). In the latter study, only two infants (0.3%) had congenital hemangiomas (CH) which are differentiated from IH on the basis of clinical behavior, pathologic features and the expression of glucose transporter-1 (GLUT-1) (21). The latter marker is expressed in virtually all IHs whereas all other vascular anomalies and malformations, including CHs, are GLUT-1 immunonegative (22,23). The study of IHs has provided opportunities to better understand the morphogenetic mechanisms of angiogenesis and vasculogenesis and their signaling pathways (24,25,26). A placenta-derived vascular stem cell has been postulated as the progenitor of IH (27,28). Immunohistochemical studies have shown that IHs express stem cell, neural crest and pericyte phenotypes (29).
There is a female prediction and a preference for the head and back region (40% to 60% of all cases) for IHs (30,31). Most IHs are solitary lesions (75% to 80%); the remainder are multifocal and there is an increased risk for visceral involvement in the presence of multiple lesions. In one study, 24 (16%) of 151 infants with five or more IHs had hepatic IHs (32). Like the skin or deep soft tissues, visceral lesions also demonstrate GLUT-1 reactivity (33).
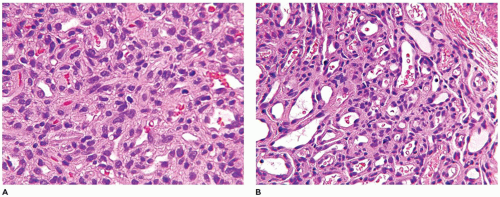
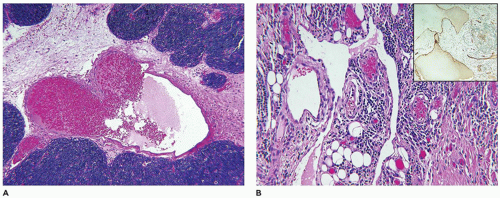
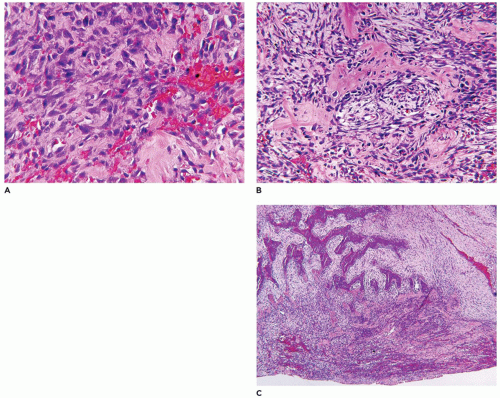
FIGURE 25-1 • Infantile hemangioma. A: The proliferative phase shows marked cellularity, mitotic figures, and a few erythrocytes to delineate the vascular spaces. B: The involution phase is characterized by readily identifiable capillary-sized vascular spaces.
The natural history of IH is reflected in the clinical and pathologic findings. Initially, there is an erythematous macular area which undergoes progressive enlargement over a period of a few months (known as the proliferating phase); this phase is succeeded by deceleration and stabilization in growth (the involutional phase) (Figure 25-1A, B). A vascular anomaly present at birth is either a CH or VM and not an IH, since IHs are only occasionally present at birth.
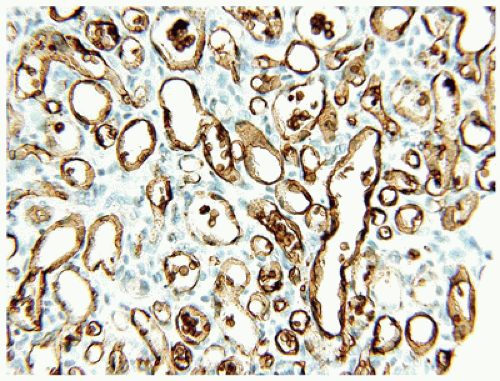
Both IHs and CHs have lobular patterns of growth so other features of the particular vascular anomaly must be applied to differentiate one from the other. During the proliferating phase of an IH, the lobules have a hypercellular appearance since the vascular lumina are small and may accommodate no more than a single erythrocyte. Both endothelial and pericytic cells surround the vascular spaces. Mitotic figures are identified without difficulty during this phase and Ki-67 nuclear proliferation antigen is strongly and diffusely expressed. The involutional phase is appreciated microscopically by the presence of open vascular spaces with flattened endothelial cells as the lobules lose their hypercellularity associated with the proliferative phase. There is relatively little fibrous stroma associated with an IH. These lesions can be quite extensive throughout the dermis and into the subcutis. As a matter of establishing the diagnosis of IH, GLUT-1 immunostaining should highlight the constituent endothelial cells (Figure 25-2). Erythrocytes helpfully serve as an internal control but not so helpfully obscure the endothelial reaction when the capillary-sized vascular spaces are filled with red cells.
Clinical involution occurs in 85% to 90% of IHs over a period of months or even longer in some instances (30). Occasionally, a resection specimen is from the region of skin that had been the site of an IH; a fully regressed IH is characterized by pallor of dermal collagen with relatively inconspicuous vessels that nonetheless retain staining for GLUT-1.
One additional note is the possibility of PHACE syndrome or association in infants with large IHs of the face; this syndrome is further characterized by extracutaneous vascular anomalies in 25% to 30% of cases (posterior fossa vascular anomalies, hemangiomas, arterial anomalies [such as aberrant subclavian artery], cardiac defects [such as aortic coarctation] and vascular anomalies of the eye) (34,35,36).
Congenital hemangioma (CH) includes two GLUT-1-negative vascular anomalies:the rapidly involuting type (RICH) and the noninvoluting congenital hemangioma (NICH) type (21,37). Though both lesions may clinically resemble the IH, they are considerably less common than the IH (only 5% to 7% of all hemangiomas). The designation “congenital” hemangioma denotes the presence of these lesions at birth rather than the usual postnatal development of IH (38,39).
FIGURE 25-2 • Infantile hemangioma. Endothelial immunostaining for GLUT-1 confirms the diagnosis of infantile hemangioma.
RICH, like the NICH, may be detected in utero by ultrasonography since both have a rapid blood flow pattern. At the time of birth, the growth of a RICH may have already peaked and involution/regression has already begun and is usually complete by two years of age. Dystrophic calcifications are present in 35% to 40% of cases (37). Like the IH, both RICH and NICH have a predilection for the head and back region (39). Consumptive coagulopathy and/or high output congestive heart failure are present in a minority of cases. Once RICH involutes, a pocket of loose skin often remains. Nasseri et al. (40) have reported a series of RICHs which only partially involuted. Within the center of the lobules, fibrosis may be evident with loss of capillaries (41). Variably sized vascular lobules are surrounded by dense fibrous stroma. Large draining vascular spaces are present within the extralobular stroma with evidence of thrombosis, hemosiderin deposition and calcifications (Figure 25-3). Both large and small vascular lobules are composed of plump endothelial cells and a cuff of pericytes. As previously noted, the RICH is GLUT-1 negative, but the nuclei are immunopositive for WT1.
NICH have usually attained their maximum growth at the time of birth, but remain clinically stationary for life (42). Like IH and RICH, NICH has a predilection for the head and neck but has a more anatomically diverse distribution than the other two anomalies and can also be found on the trunk and extremities (43). These lesions are typically solitary (39). Scattered vascular lobules with a hypercellular appearance within the dermis have some resemblance to the tufted hemangioma (TA) (Figure 25-3). The center of the lobules has a large vessel with a stellate appearance. Ectatic, thinly muscularized vessels and smaller arteries are present with proximity to the vascular lobules. The presence of these vessels may suggest a VM. Dystrophic calcification may be present, but less often than in the RICH. Also as noted, GLUT-1 is nonreactive as is D2-40 if there is concern about a TA (43). There are differences and similarities in the molecular fingerprint of IHs and CHs as reported by Picard and associates (44).
FIGURE 25-3 • Congenital hemangioma (non-involuting type) is characterized by multilobular growth pattern. Some lesions may require GLUT-1 immunostaining to rule out infantile hemangioma especially in cases without a history.
Verrucous hemangioma (VH) is another congenital or neonatal vascular anomaly whose eponymous verruciform epidermal hyperplasia has features resembling the angiokeratoma. The linear growth is like that of a linear epidermal nevus. These lesions have a predilection for the extremities where early in the course of their evolution, epidermal alterations may not be readily apparent. Arrays of capillary-sized vascular spaces are situated in the papillary dermis beneath the epidermis, as well as similarly sized vessels in the deeper dermis and subcutis with a lobular and/or diffuse growth pattern (17). Like the IH, the vessels are GLUT-1 positive (37). There are those who consider the VH as a VM, but the GLUT-1 reactivity favors a neoplastic process like the IH.
OTHER VASCULAR NEOPLASMS
Beyond the IH and CH other vascular neoplasms comprise a myriad of tumors whose combined numbers in children account for less than 5% of all cases. Included among these neoplasms are the microvenular, hobnail (targetoid), tufted and spindle cell hemangiomas (SCHs).
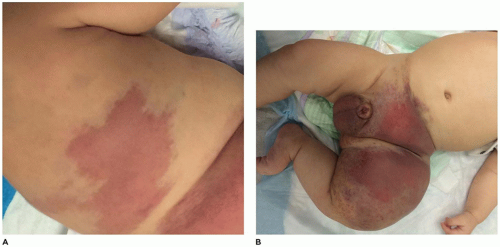
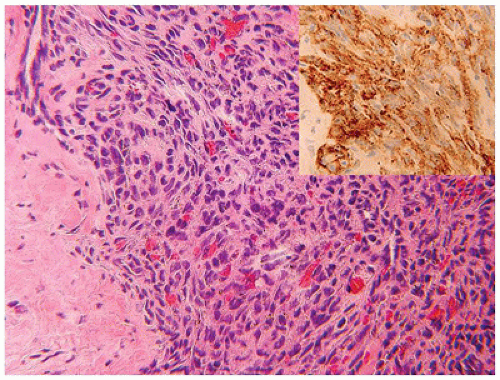
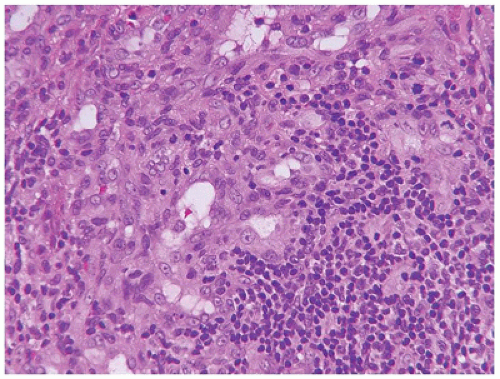
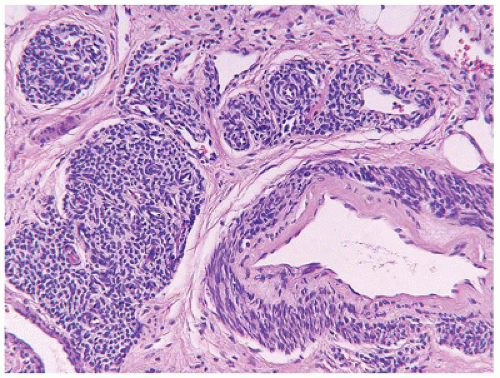
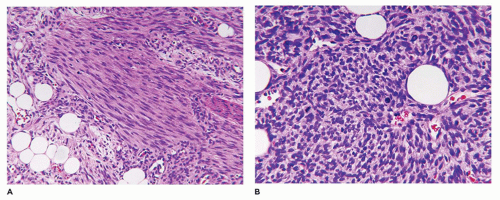
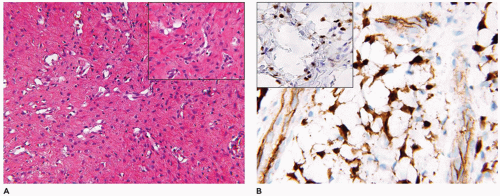
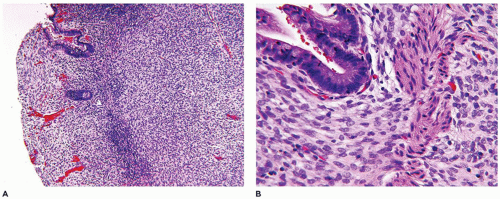
Tufted hemangioma (TA) and kaposiform hemangioendothelioma (KHE) are regarded as histogenetically related neoplasms, if not the same entity, in that both express the lymphatic endothelial marker, D2-40, and the lymphatic endothelial nuclear transcription factor, Prox 1 (45,46). Both tumors can be complicated by the eponymous coagulopathic process, Kasabach-Merritt phenomenon (KMP) or syndrome, which presents in neonates (60% of cases) or infants (47,48). A bluish purple patch or plaque on the trunk, neck or extremity with or without hypertrichosis in an infant is the clinical appearance of the TA whereas a deeper soft tissue mass in a similar anatomic distribution is the presentation of KHE (Figure 25-4). The deeper and larger KHE is more often complicated by KMP.
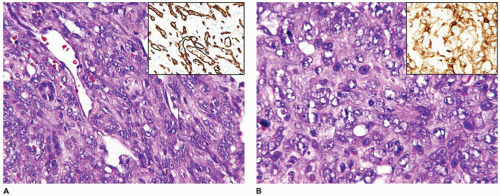
A deep punch biopsy of a cutaneous TA shows the presence of variably sized and numbered cellular nodules in the dermis and subcutis. The nodules are composed of slit-like or ovoid vascular spaces with dilated, crescent-shaped spaces at the periphery of the nodule (49). Larger nodules may have distinctly spindle cell features resembling the KHE. The endothelial cells can have plump epithelioid features and the nodules may be centrally sclerotic. In contrast, KHE is a larger mass in the deep soft tissues including skeletal muscle and growth into bone. Other sites of KHE include the retroperitoneum, mediastinum and intestinal tract. The spindle cell pattern of KHE is composed of slit-like spaces with erythrocytes and hyaline globules like Kaposi sarcoma (KS) (Figure 25-5). However, the tumor cells have a more bland appearance, mitotic activity is sparse and hemosiderin is less conspicuous in the KHE. Like KS, the tumor cells are positive for CD31 and D2-40, but unlike KS, there is absence of human herpesvirus 8 (HHV8) by IHC (Table 25-5).
FIGURE 25-4 • Kaposiform hemangioendothelioma. A: This infant presented initially with erythematous slightly raised plaques. B: Only a few weeks later, the two lesions had coalesced into the bluish mass occupying the entire thigh (Contributed by Adam Vogel, M.D., St. Louis, MO).
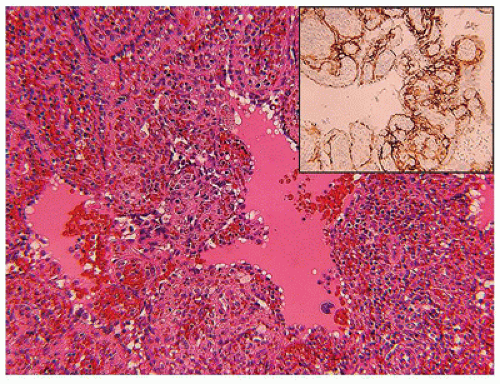
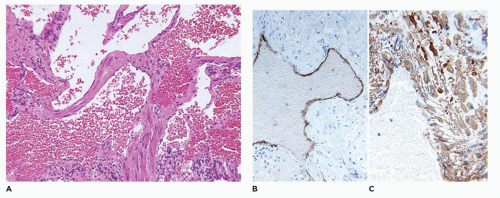
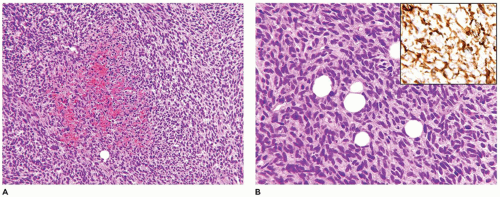
Kaposiform lymphangiomatosis (KLA) is a vascular anomaly which may also present with consumptive coagulopathy, and/or hemoptysis, pleural and pericardial effusions due to intrathoracic involvement of the mediastinum and lungs (50). The median age at diagnosis is 6.5 years which is considerably older than in the case of KHE. The spleen and bones may be involved unlike KHE (Figure 25-6A, B). Microscopically, spindle cells and lymphatic endothelial cells like those seen in KHE are present, but there are accompanying ectatic lymphatic spaces of a lymphatic malformation or “lymphangioma” (Table 25-5). A small biopsy may not show the spindle cell component. Morphologically, there are subtle distinctions from KHE, but lymphatic spaces are apparent in KLA (Figure 25-7). The overall survival is only 35% or so (50).
FIGURE 25-5 • KHE presented as multiple masses in the intestinal tract and retroperitoneum in a 7-year-old female. A: Many of the nodules are composed of compact spindle cells with interposed erythrocytes resembling KS. B: Other nodules had a more lobulated and tufted appearance. Inset: Factor VIII-related antigen immunostaining labeled the spindle cells.
TABLE 25-5 KAPOSIFORM VASCULAR LESION AND KAPOSI SARCOMA
KHE
KLA
KS
Age
Infants
Early-late childhood and few adults
Adults except lymphadenopathic form in children
Distribution
Solitary, but locally extensive, soft tissue, retroperitoneum
Epithelioid hemangioma (EH) and hemangioendothelioma (EHE) are similar appearing vascular neoplasms composed of plump endothelial cells with abundant eosinophilic cytoplasm and intracytoplasmic vacuoles representing luminal formation (Figure 25-8). Though there remains some confusion in the pathologic distinction between EH and EHE, the latter is regarded as malignant together with angiosarcoma. Even in aggregate, EH and EHE are very uncommon in children. Angiolymphoid hyperplasia with eosinophilia is one cutaneous presentation of EH; most cases present in the head and neck region (51,52,53). Erythematous papules are composed of lymphoid aggregates in the dermis with accompanying eosinophils which may obscure the small vessels lined by plump endothelial cells; these vessels have a vague lobular architecture (54). Bone, soft tissue and blood vessels are other sites of EH in children. EHE lacks the lobular architecture of EH and is composed of nests, cords and strands of epithelioid cells with vacuoles resembling signet ring cells (55) (Figure 25-9). A myxoid or myxohyaline stroma is often prominent in the background. There is an angiocentric orientation of this tumor in the soft tissues whereas those arising in the bone, liver and lung are multifocal in 50% or more of cases (56). The liver is seemingly the most common site of EHE in children, usually in the adolescent years. There is a CAMTA1-WWTR1 fusion transcript with the t(1;3) (p36.23;q25.1) translocation; the latter is found in all EHEs regardless of primary site and is not found in other vascular neoplasms (57).
FIGURE 25-6 • Kaposiform lymphangiomatosis involving the spleen and other sites in a 3-year-old female. Branching and anastomosing vascular spaces, some with a pale coagulum representing lymph. Inset: D2-40 immunostain highlights the endothelial-lined spaces.
The differential diagnosis of EH and EHE includes other epithelioid vascular and nonvascular neoplasms. One of these is the cutaneous epithelioid angiomatous nodule (CEAH) which resides in the dermis as a well circumscribed nodule with some overlapping features with EH except for the less intense inflammation in CEAH (58,59,60). This vascular lesion is seen in older children and adolescents. Epithelioid sarcoma-like hemangioendothelioma (pseudomyogenic EHE) is a keratin-positive and INI1-retained vascular neoplasm occurring in the skin, soft tissues, or bones of children and young adults (61,62,63). There is a predilection for the extremities (lower greater than upper) and multifocal lesions are present in some cases. In addition to spindle cells, the predominant cell type has rhabdoid or epithelioid features. Vascular spaces are readily apparent and eosinophils and neutrophils may be seen in the background. Local recurrence or the development of new lesions is seen in 50% to 60% of cases. This neoplasm has a t(7;19)(q22;q13) translocation representing a SERPINE1-FOSB fusion transcript (64).
FIGURE 25-7 • Kaposiform lymphangiomatosis. This bone biopsy shows a lobule of spindle cells with a resemblance to SCH and KHE. Inset: D2-40 immunostain reacts with lymphatic endothelial cells.
FIGURE 25-8 • Epithelioid hemangioma presented as a cutaneous mass on the arm of a 3-year-old male. Compact small vessels lined by epithelioid or histiocytoid endothelial cells, some with vacuolated cytoplasm and an accompanying lymphocytic infiltrate characterize this lesion.
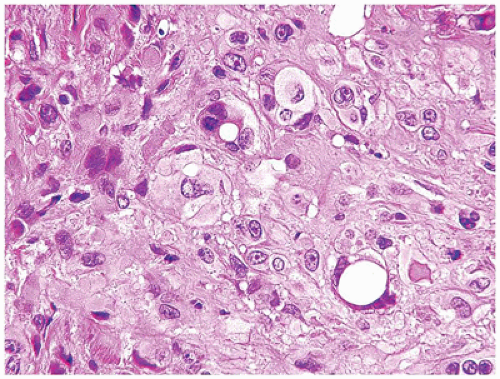
Angiosarcoma (AS) is a rare malignancy at any age let alone childhood. EHE is the low-grade variant of AS whereas epithelioid and classic ASs are both high-grade malignant neoplasms. The skin is the most common site of AS in adults, but in children, even infants, this tumor presents in the liver, spleen, and superficial and deep soft tissues (65,66) (Figure 25-10A-D). Epithelioid AS may not suggest that diagnosis initially in either a child or an adult in the absence of recognizable neoplastic vascular structures, but the presence of hemorrhagic background and eosinophils is a clue to the diagnosis. Classic AS is composed of high grade round or spindle cells and is often accompanied by a hemophagocytic background. Papillary profiles may be present creating the so-called type II pattern of infantile EHE of the liver which we and others now believe is AS. In the latter case, retiform profiles with prominent endothelial cells assume a pattern resembling the retiform EHE.
FIGURE 25-9 • Epithelioid hemangioma. This tumor contained both epithelioid cells with a moderate degree of cytologic atypia and cells with a prominent intracytoplasmic vacuole.
Retiform hemangioendothelioma (RHE) is a rare vascular neoplasm of the dermis and subcutis which presents in children as a reddish plaque or mass with infiltrative borders (67,68). Though its histologic features have been likened to the rete testis with aggregates of small vascular structures, its growth through the dermis and into the subcutis resembles AS except for the absence of marked unclear atypia and mitotic figures. The vascular channels tend not to have an infiltrative pattern, but a more arborizing one. These cases can be challenging regardless of the age of the patient. A lymphangioma-like component and small glomeruloid profiles within the vascular channels are other features to suggest the diagnosis of RHE.
Papillary intralymphatic angioendothelioma (PILA, Dabska tumor) was first recognized in children, but is also seen in adults (69). There are some overlapping features between the PILA and RHE. Variably sized vascular spaces are immunoreactive for the lymphatic marker, D2-40, and a subset contain the characteristic intraluminal papillary endothelial-lined structures. Unlike RHEs, PILAs generally do not locally recur.
Composite hemangioendothelioma (CHE) is another low grade vascular neoplasm which like several of the other tumors in this section is rare, and rarer still in children (70,71). There is a predilection for the distal extremities where the CHE has patterns of EH, RHE, SCH, cavernous hemangioma, EHE and AS-like foci (72). A sheet-like growth of epithelioid and spindle cells may appear to occupy a lymph node (71). This tumor has been observed in the setting of Maffucci syndrome (72). The polymorphous EHE is regarded as a distinct entity from CHE (73).
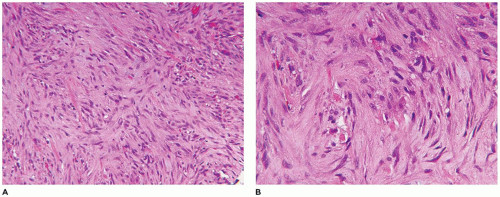
Spindle cell hemangioma (SCH, spindle cell EHE), once thought to be a low grade vascular neoplasm, is now considered a benign tumor; this tumor occurs in older children and young adults in the more superficial soft tissues of the distal extremities as a circumscribed mass (74). More than 50% of SCHs are intravascular where the pattern is a mixture of spindle cells and vascular structures with cavernous and epithelioid features (Figure 25-11A-D). Approximately 5% of cases are diagnosed in the clinical setting of Maffucci syndrome. In addition to immunoreactivity for CD31 and CD34, this tumor is also D2-40 positive (Figure 25-11D). Somatic hotspot heterozygous mutations on IDH1 (2q34) and IDH2 (15q26.1) have been detected in the enchondromas of Maffucci syndrome and Ollier disease as well as in the SCH (75,76).
FIGURE 25-10 • Angiosarcoma of the liver in a 3-year-old boy with a 15-cm hepatic mass and pulmonary nodules. A: Foci of this neoplasm have retained the pattern of an infantile hemangioma. Inset: GLUT-1 immunopositivity is present. B: Other areas show a more solid pattern of atypical polygonal cells. Inset: GLUT-1 immunopositivity is partially lost.
FIGURE 25-11 • SCH presenting on the palmar surface of the hand of a 9-year-old male. A: Low magnification of this circumscribed mass filling the dermis. B: The circumscribed mass has a variable pattern of uniform spindle cells and irregular ectatic vascular spaces. C: The spindle cells are uniform and do not display any worrisome atypia. D: The tumor is immunoreactive for CD31, CD34, and D2-40 (shown).
FIGURE 25-12 • Lobular capillary hemangioma presented on the shoulder of this young female. A: A polypoid mass is composed of lobules of capillary-sized vascular spaces. B: In the more cellular or proliferating areas, vascular spaces are difficult to appreciate, and mitotic figures are found with ease. C: The involuting areas are characterized by well-formed, patent capillaries. Some CH do not involute.
Kaposi sarcoma in children is most commonly seen as the endemic/African subtype which typically presents with regional and generalized lymphadenopathy in the absence of cutaneous lesions (77). A substantial proportion of children with KS are HIV positive, but examples are reported in children who are transplant recipients (78). As in virtually all other cases of KS, HHV-8 is positive (77) (Table 25-5).
Lobular capillary hemangioma-pyogenic granuloma is one of the most common vascular tumors in childhood whose pathogenesis as either a neoplasm or a reactive process remains somewhat ambiguous; recent studies tend to favor a reaction to tissue injury (79). This tumor occurs throughout childhood and adolescence as a rapidly evolving polypoid or pedunculated lesion, often in the head and neck region (including the oral and nasal cavity) or the finger, but has a ubiquitous distribution and may also present as multiple lesions. LCH-PG often ulcerates, bleeds and forms a crust of red cells, neutrophils and fibrin to the extent that the undergoing histologic features may be substantially obscured. Capillary-sized vascular spaces are arranged as lobules beneath the intact or ulcerated epidermis. Collarettes of epidermis wrap beneath the peripheral vascular lobules in most, but not all cases (Figure 25-12A-C). A central vessel is often present at the base of each lobule. A similar appearing lobular vascular lesion can be seen in the subcutis or within the lumen of a small blood vessel. LCH-PG seemingly evolves from a proliferative to involutional phase in a manner similar to IH. A cautionary note is that rarely KS and more often bacillary angiomatosis (BA) cases both have LCH-PG-like features. However, BA does not have the exquisite multilobulation of LCH-PG and the intervening stroma contains neutrophils and amorphous granular material representing either Bartonella henselae or B. quintana and macrophages. Both immunocompromised and immunocompetent children can develop BA (80,81).
VASCULAR MALFORMATIONS
The other major category of vascular anomalies in children is VMs which accounted for 33% of cases in a retrospective study of children with vascular lesions (19). “Low flow” VMs are composed of veins, capillaries or lymphatics in contrast to “high flow” VMs of arterial derivation. The remaining VMs are complex anomalies of veins, capillaries, arteries and lymphatics (82,83,84). Additionally, VMs occur in the settings of several syndromes (Table 25-6). The low flow VMs comprise almost 40% of all VMs whereas the isolated capillary VMs represent another 30% (19). High flow arterial-arteriolar-venous VMs constitute less than 5% of all cases. It is notable that the various syndrome-associated VMs have a distinctive morphologic composition of vessels; examples include the arteriovenous VMs in hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome), capillary malformations the macrocephaly-capillary malformation disorder and the mixed VM-connective tissue elements of the PTEN hamartoma tumor syndrome (85,86,87,88).
TABLE 25-6 REGIONAL AND DIFFUSE SYNDROMES ASSOCIATED WITH VASCULAR MALFORMATIONS
Syndrome
Malformation
Regional syndromes with slow-flow VMs
Sturge-Weber
Facial and intracranial capillary malformation, venous malformation, or AVM
Kippel-Tenaunay
Limb, trunk capillary, or venous-lymphatic malformation and overgrowth
Regional syndromes with fast-flow VMs
Parkes-Weber
Capillary AVM with overgrowth; lymphatic malformation
Diffuse syndromes with slow-flow VMs
Blue rubber bleb nevus (Bean)
Multiple venous malformations of the skin, musculoskeletal system, and GI tract
Proteus
Capillary or venous malformations, hamartomas, asymmetric limb or digit overgrowth in various body parts, lipoma, pigmented nevi
Gorham-Stout
Spontaneous osteolysis and lymphatic malformations; intercranial
Skin, mucous membrane, and GI telangiectasias; AVM of lungs, liver, brain, and spinal cord
Wyburn-Mason
AVM or AVF of brain or retina with same-segment facial malformations
Cobb
AVM or AVF of spinal cord with same-segment skin VM
AVM, arteriovenous malformation; PTEN, phosphatase and tensin homology suppressor gene; AVF, =arteriovenous fistula. From Kollipara R, Dinneen L, Rentas KE, et al. Current classification and terminology of pediatric vascular anomalies.
AJR Am J Roentgenol 2013;201(5): 1124-1135.
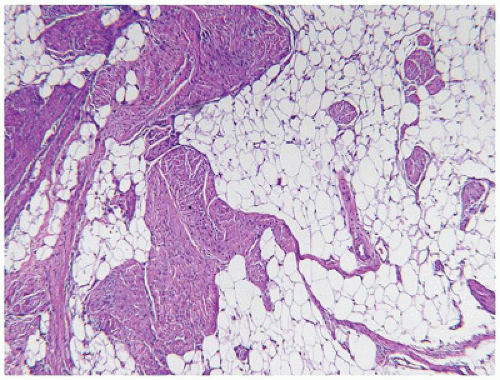
No one single description is sufficient to characterize VMs from the perspective of their morphologic features (Figure 25-13). The pure lymphatic VM, also known as a cystic hygroma or lymphangioma, is composed of thin-walled vascular structures which are empty or contain a pale coagulum (lymph) or red cells as a result of venous efflux of blood (84,85). Lymphoid nodules residing beneath lymphatic endothelium or prolapsed into the vessel lumen are seen in some but not all cases (Figure 25-14). Smooth muscle beneath endothelium may indicate the presence of veins and thus a lymphovenous malformation (Figure 25-15). Immunostaining for D2-40 is helpful in the identification of lymphatic component when it is not apparent from the microscopic examination. Prior surgery often results in a marked fibrous tissue reaction which substantially alters the appearance of the malformation; a similar change may be seen in large VMs in a site like the mediastinum.
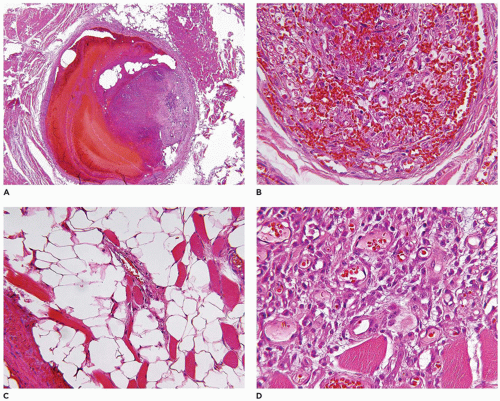
FIGURE 25-13 • VM from the posterior thigh of a 15-year-old male. This deceptively well-circumscribed mass has a range of gross findings from the presence of thrombosed vessels in various stages of formation and organization. Several foci of dystrophic calcification, mainly in vessels, are noted.
FIGURE 25-14 • Lymphatic malformation presenting as a mediastinal mass in a 3-year-old female. A: The ectatic thin-walled vascular channels are partially filled with lymph and red cells. B: These lesions dissect through and around structures in the soft tissues including the thymus. Inset: D2-40 immunostain outlines the lymphatic spaces.
Those VMs composed of veins and arteries, like the lymphatic VMs, are typically poorly circumscribed and can involve the cutaneous, subcutaneous and deep soft tissue compartments (88) (Figure 25-16). There is often an infiltrative pattern of variably sized vascular spaces filled with red cells and/or thrombi in various stages of organization. Dystrophic calcifications, metaplastic bone and adipose tissue are other findings in selected cases. Hemorrhage into the soft tissues is often followed by hemosiderin deposition and collections of hemosiderin-laden macrophages. In addition to VMs, the PTEN hamartoma, also an infiltrative process, is composed of myxoid fibrous tissue, adipose tissue, and lymphoid nodules; the hamartomatous designation for these lesions is an acknowledgment of this disorganized overgrowth of the various mesenchymal components of the soft tissues (Figure 25-17) (87).
FIGURE 25-15 • Mixed venous and lymphatic malformation presented as a mass on the chest wall of a 17-year-old female. A: A mixture of muscle associated venous structures are intermixed with thin-walled lymphatic-like spaces. B: Some of these spaces stain positively for D2-40, a lymphatic marker. C: Many of the vascular channels are associated with SMA positivity.
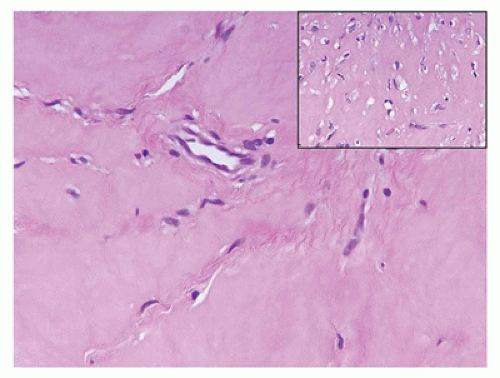
Glomus tumor (glomuvenous malformation) is regarded as a malformation of the neuromyoarterial body rather than a true neoplasm. Solitary glomus tumors (90% of cases) have a predilection for the subungual regions on the hand (88). Autosomal dominant multiple glomus tumors or glomangiomas (10% of cases) are characterized by a germline mutation in the GLMN gene on 1p22 (89). It is also of interest that there may be an increased risk of glomus tumors in the setting of neurofibromatosis type 1. Approximately 10% to 15% of solitary glomus tumors present before the age of 20 years, usually in the second decade. The glomus cell is a modified smooth muscle cell that is usually immunopositive for alpha-smooth muscle actin and vimentin, but usually negative for desmin (90). Cuffs of small uniform, basophilic cells are present beneath an intact, inconspicuous endothelium (Figure 25-18).
FIGURE 25-16 • Mixed arterial and venous VM. A: One of the larger vessels shows varying stages of thrombus formation with the more recent clot on the surface. B: This vessel has a complex network of small vascular spaces as a feature of an organizing thrombus as a single-stage process. C: Elsewhere in this malformation, small vessels infiltrate the adipose tissue. D: Small vessels infiltrate into the skeletal muscle. These various patterns were present in the gross specimen seen in Figure 25-13.
FIGURE 25-17 • PTEN hamartoma. This mass, one of several, on the shoulder of a 14-year-old female. A: Blood vessels of varying sizes and types are seen in a background of loose, somewhat immature-appearing mesenchyme. B: Another focus shows the myxoid stroma and scattered large and small blood vessels.
FIGURE 25-18 • Glomangioma (glomuvenular malformation) in a 20-year-old female presented as paraspinal and retroperitoneal masses. The vascular spaces are accompanied by a circumferential population of small, basophilic-appearing cells beneath the endothelium.
FIBROUS, MYOFIBROBLASTIC, AND PERICYTIC TUMORS
This section is focused on the category of non-neoplastic and neoplastic entities which have in common a spindle cell with the morphologic and phenotypic features of a fibroblast, and/or a transitional type mesenchymal cell, the myofibroblast, with the composite attributes of a fibroblast and smooth muscle cell which has a smooth muscle phenotype and vascular perithelial localization (91,92). In the setting of one of the unique fibrous tumors of childhood, infantile myofibromatosis-myofibroma, the proliferating subintimal myofibroblasts have the capacity to differentiate into cells with the morphology and immunophenotype of pericytes with contractile attributes of smooth muscle.
SCAR AND KELOID
Scars are reactive fibrous and myofibroblastic proliferations that occur in all tissue types and organs (brain excepted with its reactive gliosis) and are a programmed process of repair. It is not always clear as to the cause when a scar is submitted as a “tumor” in a child. Morphologically, the myofibroblasts and fibroblasts can acquire a significant degree of atypia, especially in a radiation field or site of inflammation which can be a source of concern about its benign or malignant nature.
Keloids and hypertrophic scars are similar in many respects with the formation of nodules of reactive fibroblasts in the dermis, whose presence has obliterated or replaced the normal microanatomy; both are regarded as manifestations of abnormal wound healing (93). Both processes may have a predilection in some families and are more common in individuals of African descent (94). The ear lobe is a particular site of predilection for keloids in young women after ear piercing. Keloids are additionally characterized by groups of thickened, intensely eosinophilic bundles of collagen (Figure 25-19). Similar bundles of collagen may be seen in a desmoid tumor, mesenteric fibromatosis, and NF. Both COX-1 and COX-2 are important mediators in the pathogenesis of abnormal wound healing (95,96,97).
FIGURE 25-19 • Keloid presented as a mass in the posterior auricular region of a 5-year-old female. Dense acellular bundles of collagen are separated by fibroblasts.
FIBROBLASTIC AND MYOFIBROBLASTIC TUMORS
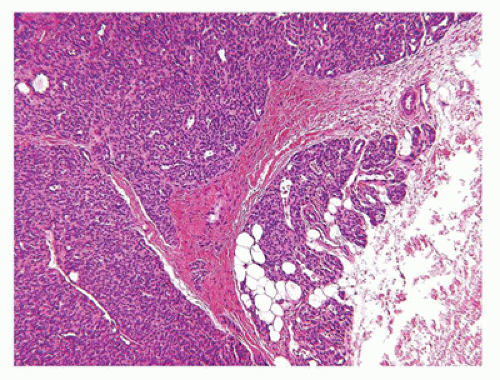
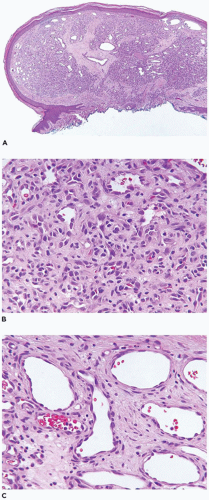
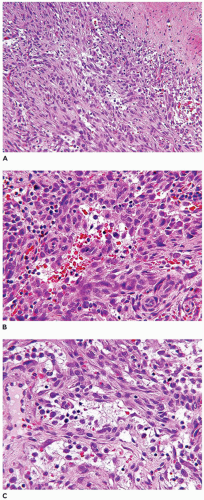
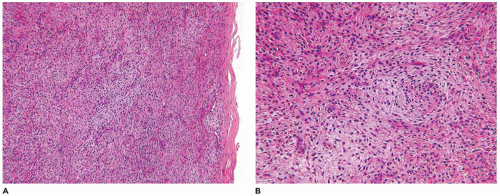
Nodular fasciitis (NF) is now regarded as a self-limiting neoplasm rather than a reactive process and is classified among fibrous/myofibroblastic tumors (5). It is unclear at the present whether the other “pseudosarcomas” are neoplastic in the same sense as NF. NF remains a problematic lesion in its differentiation from a true sarcoma (98). It is generally underappreciated that NF occurs in children with some frequency including very young children (99). One of the more dramatic examples of a fasciitis in infancy is cranial fasciitis presenting as a large mass with compression of the underlying brain in some cases (100). In our experience, approximately 40% of all cases of NF present in the first two decades, particularly in the first decade of life, as a mass with a predilection for the head and neck region in over 50% of our cases where involvement of the parotid gland is seen (101) (Table 25-7). The subcutis of the extremity (upper more common than lower) and chest wall are the more common presenting sites of NF in older children and young adults. There are also examples of intravascular and intra-articular NF (102). In the head and neck region of children, ERMS may arise in the differential diagnosis. The myxoid background and mitotic figures are the findings of concern in a NF. The subcutis is the tissue level of origin for most NFs, followed by the fascia, lower dermis and skeletal muscle. Grossly, a circumscribed, nonencapsulated nodule, measuring 3 cm or less in most cases, has a glistening mucoid appearance. The periphery has nodular contours with an abrupt transition to the adjacent soft tissues. Several histologic patterns coexist in the nodule with dense, spindle cell areas forming short fascicles adjacent to less cellular foci with separation of the spindle cells by mucoid-myxoid extracellular material (so-called tissue culture pattern) and transitional areas with both patterns (Figure 25-20). Mitotic figures are readily identified with some nuclear atypia (absent atypical mitotic figures and anaplasia). Microcysts with mucin and a variable number of histiocytes, foci of interstitial hemorrhage (so-called “extravasated red cells”), and scattered inflammatory cells in the background are the constellation of histologic features (Figure 25-21). Scattered multinucleated cells and a storiform-like pattern may suggest a fibrohistiocytic proliferation. The compact spindle cell proliferation with mitotic figures serves to raise concern for “fibrosarcoma” or leiomyosarcoma. Immature, often stellate fibroblasts in the tissue culture-like foci are the features most like an ERMS. Metaplastic bone is seen infrequently. The myofibroblasts of NF express vimentin and SMA but desmin, h-caldesmon, myoD1, and myogenin are all nonreactive by IHC. In general, NF is regarded as a nonrecurring process and with the potential for spontaneous regression. The difficulty in the differentiation of cranial fasciitis from a fibromatosis is presented in a study reporting β-catenin expression in the nuclei of a putative recurring cranial fasciitis (100). The USP6 is the fusion partner of MYH9 gene to form the t(17; 22) (p13; q13) translocation in over 90% of cases (103). Aneurysmal bone cyst (ABC) also has a USP6 translocation, but with different fusion partners than NF. The stroma of an ABC has many similarities to NF. Proliferative fasciitis and myositis are regarded as related entities in a morphologic sense to the NF (104,105). There is no evidence to date that the latter two lesions have a similar translocation as NF. The epithelioid cells in proliferative fasciitis can be mistaken for rhabdoid cells (105).
TABLE 25-7 NODULAR FASCIITIS PRESENTING IN FIRST TWO DECADES OF LIFEa
aCases of NF from the Lauren V. Ackerman Laboratory of Surgical Pathology, St. Louis Children’s Hospital, Washington University Medical Center, St. Louis, MO. between the years of 1989 and 2014.
b Includes cases from nasal cavity, paranasal sinus, oral cavity and lip.
m, month; y, year; m, male; f, female.
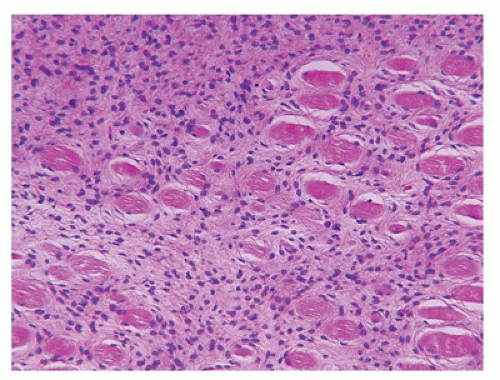
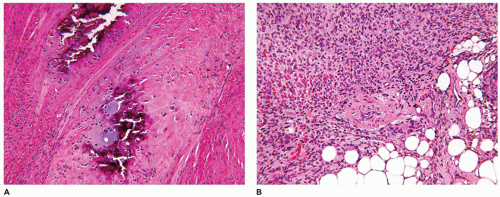
Myositis ossificans (MO), as the solitary (circumscripta), sporadically occurring soft-tissue mass or the multifocal fibrodysplasia ossificans progressiva (FOP), has its own potential for diagnostic miscues. Most cases of MO may or may not be accompanied by a history of trauma to possibly explain the male predominance (106). It is seen uncommonly in the first decade of life and more often in later childhood or adolescence in which case there may be a history of incidental or organized (sports) blunt trauma. The classic presentation is a circumscribed intramuscular mass, or alternatively the formation of a parosteal, calcified mass, or a mass attached to the surface of the bone by a pedicle. The sites of predilection are the thigh, buttock, and abdominal wall (107). In terms of size, MO can measure in excess of 10 to 15 cm. The inner portion of the three-zone mass is composed of plump spindle and polygonal myofibroblasts, blood vessels and histiocytes, which are surrounded by a zone of immature osteoid and an outer shell of mature bone (Figure 25-22). The diagnostic trap is set if a biopsy is obtained from the central, proliferating zone. Despite the initial concern for the presence of marked cellularity and some degree of atypia in a biopsy or excision, the paucity of mitotic figures, especially atypical mitoses, and the lack of anaplasia should prompt the likelihood of at least a nonmalignant process.
FIGURE 25-20 • Nodular faciitis in a 10-year-old female presented as a 2-cm mass in the posterior triangle of the neck. A: The abrupt interface exists between the spindle cells and the adjacent nonlesional collagen. Note the presence of interstitial hemorrhage. B: Intersecting fascicles and nodules of loosely arranged spindle cells and interstitial mucin are some of the characteristic features. The nuclei failed to immunostain for β-catenin. C: The presence of multinucleated cells may cause confusion with a fibrohistiocytic lesion.
FIGURE 25-21 • Nodular fasciitis presenting in the periorbital region of a 4-year-old female. A: Note the circumscribed periphery of the mass from the adjacent soft tissues and without infiltration as in the case of fibromatosis. B: Red cell extravasation is present in the interstitium. C: Several microcysts with histiocytes and mucin in the background are useful clues to the diagnosis.
Heterotopic ossification with fibro-osseous features has been reported in the auditory canal of young individuals. Cutaneous osteoma occurs sporadically or may be a manifestation of Albright hereditary osteodystrophy (AHO) or pseudohypoparathyroidism type Ia with or without the AHO phenotype. There are inactivating mutations of the GNAS gene (20q13).
FOP is an autosomal dominant disorder which is characterized by the progressive transformation of soft tissues and skeletal muscle to heterotopic bone (108). The mutation has been mapped to chromosome 2q23-24, the site of activin A type I receptor/activin-like kinase 2 (ACVR1/ALK2), a bone morphogenetic protein type I receptor. In addition to the characteristic great toe malformations, there is the development of soft-tissue swelling or masses on the back, which are described as “spreading” through the subcutaneous tissues and deeper. A biopsy reveals a spindle cell and myxoid transformation of the subcutis with a resemblance to infantile subcutaneous fibromatosis or lipofibromatosis and even more so to NF. A biopsy site may enlarge due to metaplastic ossification, which appears to accelerate in foci of trauma.
FIGURE 25-22 • MO presenting as a soft tissue mass in the posterior neck of a 10-year-old male. A: The center of this mass is composed of compact spindle cells with some nuclear atypia but in the absence of atypical mitotic figures. B: The transition zone is between the central spindle cells and osteoid formation. C: The peripheral zone is represented by the active new bone formation.
Other forms of so-called pseudosarcomatous proliferations of the soft tissues and periosteum are florid reactive periostitis, fibrous pseudotumor of the digit, a form of localized MO, and bizarre parosteal osteochondromatous proliferation (Nora lesion) (107). These various lesions are problematic if the specimen is a biopsy without adequate clinical information and characterization of the imaging features. The theme is again an atypical histology which is not accompanied by overtly malignant features, as discussed in the previous sections on NF and MO, in particular as it relates to the absence of atypical mitoses and anaplasia.
Fibroblastic-myofibroblastic tumors of childhood with some exceptions are recognized in the first 5 years of life and many at or before 2 years of age (109). These fibrous tumors of childhood or juvenile fibromatoses include the following: fibromatosis colli, myofibroma-myofibromatosis, fibrous hamartoma of infancy (FHI), inclusion body fibromatosis (infantile digital fibroma), infantile fibromatosis (lipofibromatosis and other subtypes), Gardner-nuchal fibroma, juvenile aponeurotic fibroma and nasopharyngeal angiofibroma (109). Palmar and plantar fibromatoses (superficial fibromatosis) and desmoid-type fibromatosis (deep fibromatosis) are seen in all age groups. Desmoid-type fibromatosis or desmoid tumor, Gardner fibroma, and nasopharyngeal angiofibroma are known manifestations of familial adenomatous polyposis (FAP) syndrome including Gardner syndrome (110).
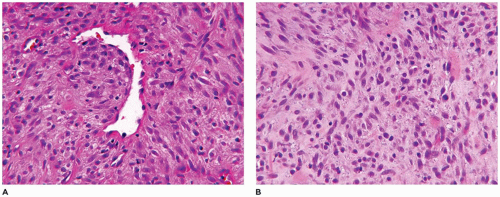
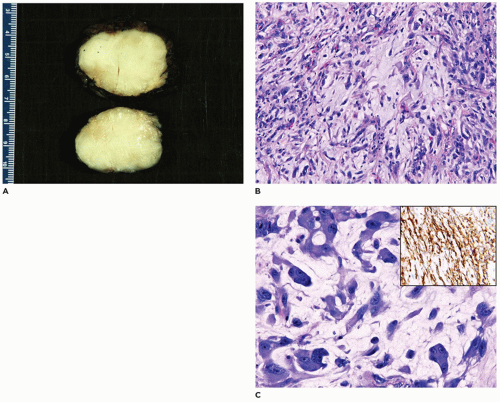
Infantile myofibroma (myofibromatosis), the most common of various fibrous tumors of childhood, accounts for 20% to 25% of all cases (109). A solitary cutaneous or subcutaneous nodule (90% of cases) measures less than 3 cm in most cases, presents in the first 5 years of life and may be noted at birth and has a predilection for the head and neck region (40% to 60% of cases) followed by the trunk and extremities (111). However, the bone and various organs including the brain, dura, liver, intestinal tract, lung, and testicle are some of the other less common solitary sites, but all of these locations may be involved in congenital generalized myofibromatosis (CGM). The cardiac fibroma occurring in infancy may be a related lesion. Virtually no site is immune to the development of a myofibroma. Multifocal lesions, usually restricted to the skin-subcutis and/or bone, are seen in 5% to 8% of cases, and in 1% to 2% of cases, there are more widespread skin, soft tissue, and visceral lesions, typically recognized in an infant less than 6 months old, comprising the entity of CGM. The clinical outcome is poor in these infants with CGM because of pulmonary venous occlusion by the formation of intravascular myofibromas. On the other hand, solitary lesions are known to undergo spontaneous regression. Another aspect of infantile myofibroma is its familial presentation with autosomal dominant or recessive inheritance with germline mutations in PDGFRB, (5q33.1) or NOTCH 3 (19p13.2-p13.1).
FIGURE 25-23 • Infantile myofibroma (myofibromatosis) presented as deep mass in the posterior neck of a 3-month-old female. A: The sharply demarcated mass measuring 3.0 cm is composed of uniform spindle cells in a pale eosinophilic stroma. B: Sweeping arrays of spindle cells are present in a fibrohyaline stroma with a small vascular space that has been compressed by spindle cells.
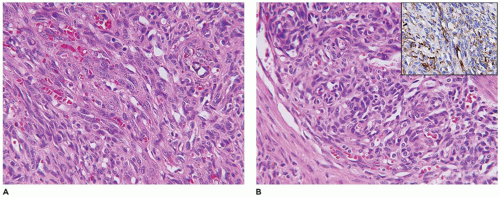
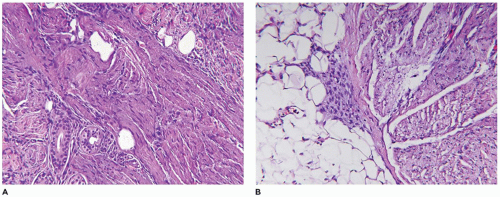
A firm nodular non-encapsulated mass measuring 1 to 3 cm in greatest dimension may also be accompanied by calcifications, cysts, and central hemorrhage with a microscopic pattern of a hemangiopericytoma (HPC) with coagulative type necrosis. The cellularity is most apparent toward the periphery where the compact spindle cells are arranged in short fascicles or within hyaline-myxoid, almost chondroid-appearing stroma, which separates or largely replaces the spindle cells (Figure 25-23). If the nodule is located in the dermis, there are often multiple discrete nodules with normal intervening cutaneous structures and dermal collagen and if in the subcutis, there is overgrowth and entrapment of fat. A more infiltrative pattern may be seen in the dermis, but the small, SMA-positive nodules are best seen in the superficial dermis. The myofibroma does not have the infiltrative growth of a desmoid-type fibromatosis but rather an expansile type growth. At the periphery of some nodules, a compressed vessel is a hallmark feature of the myofibroma and can be identified in many cases (Figure 25-24). The associated HPC-like pattern can be dominant in some tumors with only peripheral, nodular myofibromatous foci (Figure 25-25). Central degeneration without overt necrosis is yet another feature. Cells within the myofibromatous pattern are immunopositive for SMA whereas those in the HPC-like foci are positive for CD34. In addition to the HPC-like areas, dense spindle cell foci can simulate congenital infantile fibrosarcoma (CIFS) but without the potential implications nor canonical translocation of the latter tumor (112) (Figure 25-26). Local recurrence is seen in less than 10% of cases. It should be noted that the myofibroma is seen in older children and even adults.
FIGURE 25-24 • Infantile myofibroma (myofibromatosis) presented as a soft tissue mass in the neck of a 3-month-old boy. A: The smaller nodules of spindle cells are associated with a compressed vessel at the periphery that is useful in the recognition of a myofibroma. B: Other fields are composed of fascicles and nodules with a fibromyxoid appearance.
FIGURE 25-25 • Infantile myofibroma (myofibromatosis) presented as a mass in the groin of a 4-month-old male. A: This tumor has a mixed pattern of myofibroma and HPC which in this field has the former features. B: The HPC areas are usually present centrally with more ovoid cells surrounding small, clefted vascular spaces. C: Immunohistochemical staining for SMA highlights the myofibromatous pattern without reactivity in the contiguous HPC-like foci. D: A contrasting pattern of immunoreactivity for CD34 is seen in the CD34-positive HPC-like foci and absence of staining in myofibromatous focus.
The differential diagnosis of myofibroma includes NF, not surprisingly given their shared histogenesis (113,114). Whereas NF blends with the adjacent soft tissues, there is a more abrupt peripheral margin in the myofibroma. The vasculocentric lesions with intraluminal spindle cells are diagnostic of myofibroma. Yet another neoplasm is the low grade myofibroblastic sarcoma with its subtle nuclear atypia and low mitotic activity (rarely seen in myofibroma) (113). A biopsy of a low grade myofibroblastic sarcoma may not reveal the entire range of the pathologic findings.
The myofibroblast and pericyte coexist in the myopericytoma (MPC), a tumor of skin and soft tissue, which has some resemblance to infantile myofibroma-myofibromatosis (114,115). However, MPC occurs in older children and adolescents but mainly in adults. The nodular pattern of MPC consists of small vessels surrounded by concentric collarettes of spindle cells which has a resemblance to the metanephric stromal tumor of the kidney. There is a malignant counterpart to MPC with the perivascular pattern, but by tumor cells with highly atypical features.
Solitary fibrous tumor (SFT), initially reported as a pleural based tumor, is now recognized in many extrapleural sites including the skin, soft tissue, dura and kidney (116,117). HPC presenting in older children and adults is regarded as a related neoplasm to SFT but separate from infantile HPC as part of the infantile myofibroma-myofibromatosis spectrum (118). SFT is generally a spindle cell tumor, but there is a myxoid variant (Figure 25-27). Both SFT and HPC are immunoreactive for CD34 like DFSP and a number of other spindle cell neoplasms (119). Like SS, SFT is also positive for CD99. STAT6 nuclear positivity in the SFT is a specific marker for this tumor which reflects the NAB2-STAT6 fusion gene (120).
FIGURE 25-26 • Infantile myofibroma (myofibromatosis) presented in a 7-day-old female as a mass on the back. A: One of the two patterns in this tumor includes uniform spindle cells in a pale, eosinophilic background. B: Other foci are more hypercellular and mitotically active with a resemblance to CIFS. Despite the similarities, a t(12;15) is not found in these worrisome foci.
Infantile fibromatosis includes three pathologic patterns based in part on the level of tissue involvement: subcutis with the alternative designation of lipofibromatosis; skeletal muscle with diffuse infiltration by immature appearing spindle cells; and desmoid-type fibromatosis without few specific microscopic features to differentiate it from a desmoid tumor in an older child or adolescent except for the more cellular, less collagenized background in some cases (121).
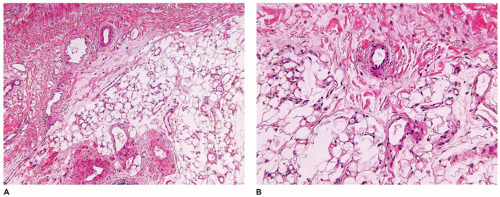
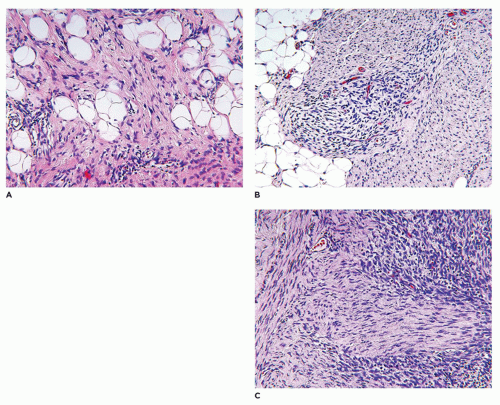
Infantile subcutaneous fibromatosis (ISF, lipofibromatosis) accounts for approximately 5% to 10% of fibrous tumors of childhood and has a predilection for the distal extremities, though it may also occur on the trunk and head and neck region (122). The growth pattern of variably dense spindle cells with a collagenous background extends along and around the interlobular septa of the subcutaneous fat and is not well circumscribed at least pathologically (Figure 25-28). There is partial overgrowth of the fat by the spindle cells with some resemblance to DFSP, diffuse neurofibroma (NF), congenital melanocytic nevus, and FHI. In fact, a fibrous-appearing DFSP should be considered in the differential diagnosis even in a young child. CD34 expression may be present focally at the periphery in the ISF (typically diffuse in DFSP), but the spindle cells of ISF are variably positive for SMA (usually negative in DFSP). FHI in the absence of the nodules of immature mesenchyme has a virtually identical pattern through the subcutis as ISF. ISF is known to recur which is not surprising, given the diffuse and infiltrative growth and frequent incomplete surgical resection. Infiltration into the fascia and muscle is uncommon.
FIGURE 25-27 • SFT from the flank of a 14-year-old male. A: The tumor has a network of regularly spaced delicate branching vessels and spindle cells in a uniform myxoid background. B: The ovoid to spindle-shaped cells have bland features. Inset: CD34 immunostain and CD99 (not shown) are positive in the vessels, and spindle cells (see Int J Surg Pathol 2013;21:358-362).
Diffuse pattern of infantile fibromatosis is recognized as an infiltrating tumor involving skeletal muscle, especially in the head and neck region and often the tongue as well as other sites (109,121). Immature spindle cells sweep through the interstitium of the muscle separating in the absence of complete destruction of the bundles of skeletal muscle (109) (Figure 25-29). Because of the relatively immature appearance of the tumor cells, fetal rhabdomyoma (FRM) or ERMS may be considered in the differential diagnosis. Appropriate immunohistochemical stains for myogenin should resolve the dilemma since the latter is only expressed in rhabdomyoblasts; however, myogenin may be expressed in regenerating skeletal muscle to create some confusion with ERMS.
Complete surgical resection is complicated by the morbidity of tumor location and its diffuse growth.
FIGURE 25-28 • Infantile subcutaneous fibromatosis (lipofibromatosis) in this 3-day-old boy in the head and neck region shows a proliferation of immature-appearing spindle cells within the subcutaneous fat. The entrapment rather than overgrowth distinguishes this fibromatosis from DFSP-GCF which is also seen in infancy.
Classic desmoid-type fibromatosis infrequently occurs in infants but its fibrous, spindle cell pattern with irregular infiltration and replacement of skeletal muscle along the infiltrating borders are identical to the desmoid tumors in older individuals. Any one of the infantile fibromatoses can involve the lower dermis, whereas involvement of the deep soft tissues including the skeletal muscle is confined to the diffuse and desmoid types (123).
Fibrous hamartoma of infancy (FHI), another unique fibrous tumor of childhood, accounts for no more than 5% of all such neoplasms (109). This tumor occurs predominantly in the first 2 to 3 years of life—even in slightly older children—but most commonly in the first few months where the trunk (60% to 65%), axilla, inguinal, and perineal region (vulva and scrotum), and extremities are the sites of predilection in descending order (109,124,125,126). The subcutaneous, poorly circumscribed fibrofatty tumor generally measures less than 5 cm. It shares many of the same gross and microscopic features with the ISF except for the small nodules of immature, spindled mesenchymal cells in a pale basophilic background (Figure 25-30). These may be found as isolated structures in the fat or along the periphery or within bundles of more mature-appearing spindle cells. Focal extension may be found into the dermis where secondary changes in the eccrine sweat glands have been observed (127). Pseudoangiomatous foci have been described with accompanying bcl-2 reactivity (126). Both smooth muscle actin and CD34 are expressed in the more fibrous areas of a FHI (125). If the lobules of fat have an immature appearance, lipoblastoma (LPB) may arise in the differential diagnosis. The local recurrence rate is only 10% to 15%, which is low in light of the fact that FHI is incompletely resected in most cases.
FIGURE 25-29 • Infantile fibromatosis of the diffuse type presented on the upper back of a 4-month-old boy. The loosely arrayed immature spindle cells are infiltrating through the skeletal muscle rather than its destructive overgrowth as in desmoid fibromatosis.
FIGURE 25-30 • FHI presented in the axillary region of a 4-month-old boy. A: The pattern of subcutaneous infiltration by bland-appearing spindle cells resembles ISF. B: The presence of small bundles of immature spindle cells at the periphery or within the midst of the more mature fibroblasts is the diagnostic feature.
FIGURE 25-31 • Inclusion body fibromatosis (infantile digital fibroma) presented on the fifth toe of a 7-month-old female. A: The dense, relatively hypocellular spindle cell proliferation has effaced the dermis. B: Trichrome stain demonstrates uniform pattern of collagen deposition. C: Paranuclear eosinophilic bodies are best seen at higher magnification. D: These filamentous bodies of actin are better demonstrated in the trichrome stain.
Inclusion body fibromatosis (infantile digital fibroma) presents on the lateral and/or dorsal aspect of a finger and/or toe, though usually sparing the thumb and great toe, as a firm nodule(s) in an infant or child 5 years of age or less at diagnosis (128). Multiple digits are involved in 25% to 30% of cases. This tumor measures 1 to 2 cm in most cases and has a uniform white, fibrous appearance similar to a desmoid-type fibromatosis. The dermis is commonly effaced by a uniform spindle cell proliferation, forming short fascicles, and with a prominent collagenous background entrapping isolated hair follicles or sweat glands (129) (Figure 25-31). Confluent, contiguous extension into the subcutis is associated with overgrowth of fat with features resembling macrodactyly. There is a microscopic resemblance to the desmoid-type fibromatosis, except for the presence of eosinophilic, paranuclear inclusions in variable numbers; these inclusions are usually more readily identified in a trichrome stain. The eosinophilic inclusions are pathognomonic of this entity when present in a digital lesion, but the inclusions have also been observed in rare extradigital fibroblastic lesions and in fibroepithelial tumors of the breast (130). The infiltrative growth around and through neurovascular structures in the digit limits complete resection in most cases, which accounts for a local recurrence rate in excess of 50% (131). There are uncommon examples of spontaneous regression (132).
Dermatomyofibroma is described in children between the ages of 2 and 16 years as a plaque or nodule, typically in the neck or upper trunk (133). A spindle cell proliferation is found in the lower dermis and subcutis with a pattern which tends to parallel the overlying epidermis (134). Smooth muscle actin is commonly positive, but CD34 positivity is seen which promotes diagnostic confusion with DFSP and the fibroblastic connective tissue nevus (135).
FIGURE 25-32 • Composite fibrous tumor in a 1-year-old male who presented with a mass in the arm, and then developed masses in the paraspinal and retroperitoneal regions. A: The tumor has areas of ISF. B: This is a focus resembling FHI. C: A third pattern with the features of myofibroma and CIFS.
A small subset of fibroblastic-myofibroblastic tumors, typically presenting in the first 2 years of life, demonstrates a collage of more than one histologic pattern of the various fibrous tumors of childhood (composite fibrous tumor). The most common example is the infantile myofibromatosis— HPC with concurrent patterns of both which is regarded as a spectrum phenomenon in the infantile myofibroma. Other combinations are the infantile subcutaneous fibromatosis with CIFS-like foci and infantile myofibroma (112). These tumors demonstrate the morphologic plasticity of the fibroblast-myofibroblast and its capacity to simultaneously express itself as several microscopic patterns and in a sense reflects the relationship of these seemingly disparate fibrous tumors of childhood (Figure 25-32). We have seen examples of composite fibrous tumor behave in the fashion of multifocal or generalized infantile myofibromatosis.
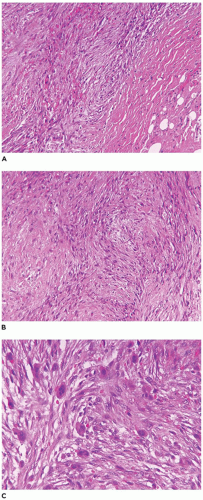
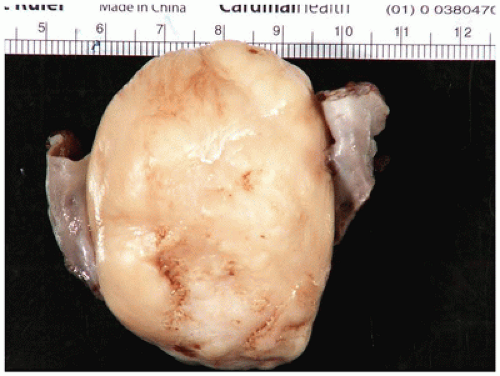
Desmoid-type fibromatosis (desmoid tumor, musculoaponeurotic fibromatosis) is the most common fibrous neoplasm presenting in the first two decades of life (60% to 70% of all fibrous tumors) and occurring throughout childhood and adolescence with a bimodal age distribution in the first 2 years and later in older children (3,109,136). The extremities (including brachial plexus) (40%) and trunk (35% to 40%) are the sites of predilection in older children, but these tumors are also seen in the head and neck (15% to 20%) and abdominal (mesentery and pelvis) sites (10%). Desmoid tumors arising in the shoulder-axilla or glutealthigh region have a local recurrence rate of 30% or greater (136,137). Most tumors occur sporadically (90% to 97% of cases), but there is an association with Gardner syndrome— FAP in less than 5% of cases; there is an increased risk for Gardner syndrome in those with FAP who have mutations between exons 1310 and 2011 (138).
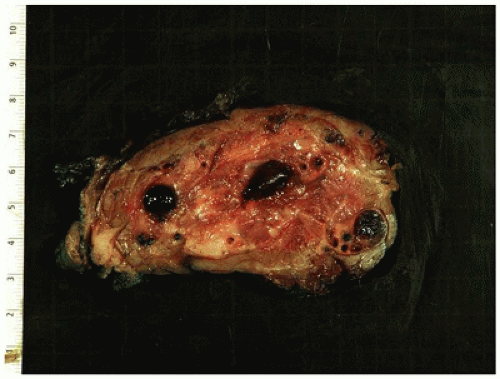
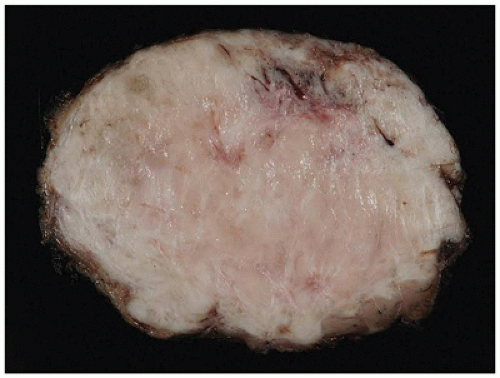
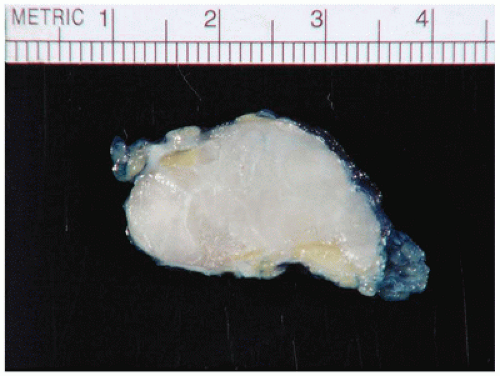
FIGURE 25-33 • Desmoid fibromatosis (desmoid tumor) presented as a deep soft tissue mass in the posterior thigh of a 15-year-old female. The cut surface of this 12-cm circumscribed mass has a tan-white trabecular appearance. Note the pushing growth into the skeletal muscle at the periphery.
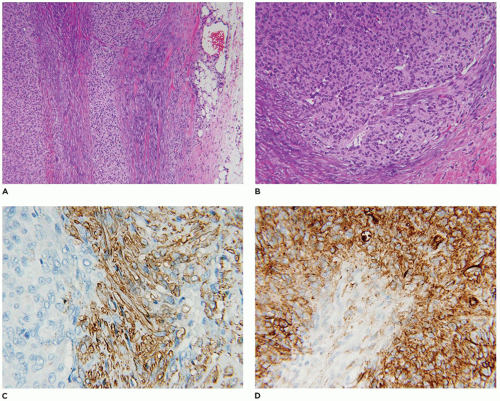
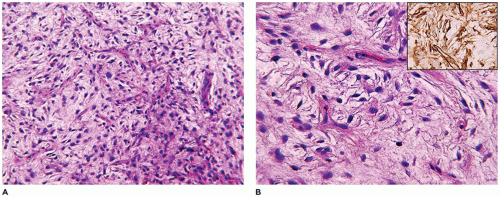
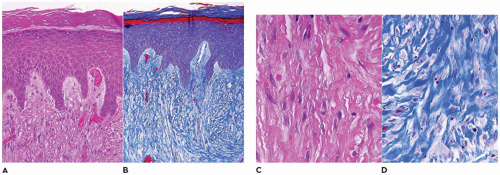
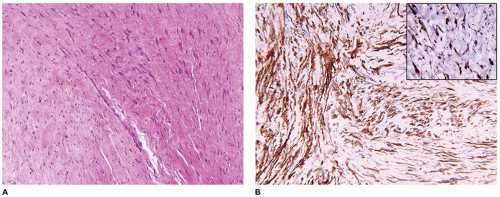
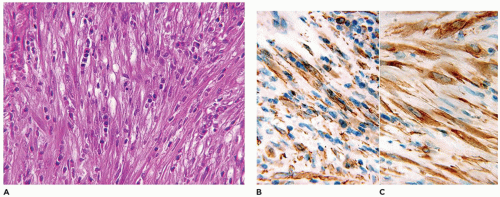
A small incisional or needle biopsy can be challenging since other non-neoplastic and neoplastic fibrous proliferations are considerations in the differential diagnosis given the limitations of a small specimen. Could this rather bland fibrous proliferation represent a scar or low grade fibromyxoid sarcoma (LGFS)? An operative resection yields a gray-white mass with a uniform mucoid to trabeculated appearance whose dimensions range from a few centimeters to >10 cm (Figure 25-33). When skeletal muscle is present at the periphery of the resection, irregular infiltration by the usually bland spindle cell proliferation into the muscle is a common feature. The periphery of the mass should be tattooed with India ink (or other appropriate dye) to document the invariably positive surgical margins. Fascicles of variably dense spindle cells or a loosely organized pattern of spindle cells are accompanied by a pale, myxoid to edematous or more collagenized background. The spindle cells may have the features of mature fibroblasts or display variation in the size and configuration of the stromal cells to reflect their less mature, more myofibroblastic features, which is manifested by cytoplasmic immunopositivity for SMA and nuclear positivity for β-catenin (Figure 25-34). Mitotic figures can be identified among the myofibroblasts but atypical mitotic figures should be viewed with wariness. A myxoid background, proliferating myofibroblasts, some interstitial hemorrhage and edema portray a more NF-like appearance. The infiltrative margins rather than peripheral nodularity characterize the desmoid tumor in contrast to NF and LGFS. The same infiltrative pattern of spindle cells into infant skeletal muscle can risk the diagnosis of spindle cell rhabdomyosarcoma. Scattered lymphoid nodules at the interface with the surrounding normal soft tissues also usefully distinguish a desmoid tumor from other fibrous proliferations. There is little to differentiate a recurrent desmoid tumor from the newly diagnosed tumor except for the findings of earlier surgery including dense scarring, foreign body giant cells, and a more circumscribed margin than in the primary tumor. Immunostaining for β-catenin is helpful in those cases when there is uncertainty whether the particular fibrous tumor is a desmoid or not; there is diffuse nuclear positivity in the desmoid tumor (139,140). There is also the disputed role of β-catenin as a prognostic marker for recurrence (141,142).
FIGURE 25-34 • Desmoid fibromatosis (desmoid tumor) presented in the posterior thigh of a 15-year-old female. A: A bland proliferation of fibroblasts is seen in a nonhomogeneous collagenous background. B: The fibroblasts maintain their myofibroblastic phenotype with immunostaining for SMA. Inset: Most desmoids express nuclear β-catenin by IHC.
The local recurrence of the desmoid tumor is 35% to 70%, which has given rise to reconsideration as to the approach in follow-up care. Some have advocated a “wait and see” approach since positive surgical margins, though predictive of recurrence, are not always associated with recurrent disease (136,137,143,144,145). Some have even called into question the desirability of primary surgery in those cases with the likelihood of serious functional or cosmetic morbidity in a child (146).
The relationship of the desmoid tumor to FAP has provided the opportunity to understand some of the molecular pathology of this neoplasm. Sporadic desmoid tumors have somatic mutations in the β-catenin gene (CTNNB1 on 3p21), which regulates the Wnt signaling pathway whereas the FAP-associated desmoids have an APC (5q22) germline mutation (138). Nuclear expression of β-catenin is a useful marker to differentiate the sporadic and familial desmoid tumors from other fibrous tumors, but some of the other fibrous tumors may also have nuclear positivity for β-catenin so that it is not absolutely specific for desmoid tumors. Nonetheless, greater than 80% of desmoid tumors express nuclear β-catenin.
Gardner-nuchal fibroma is a distinctive paucicellular, densely collagenized mass presenting in the first decade of life with a predilection for the posterior truncal-paraspinal region (147,148). Other sites of involvement include the head and neck and extremities. Approximately 70% of affected individuals have a family history of FAP or represent a new mutation. The tumor is poorly circumscribed with a plaque-like growth in the subcutis or deeper soft tissues, and there is overgrowth of the subcutis with entrapment of adipose tissue. It can be difficult to distinguish between the peripheral margins of a fibroma and the normal fibrous connective tissues. A desmoid tumor may accompany a fibroma or evolve from a recurrent fibroma. There is nuclear positivity for β-catenin but in a less consistent fashion than in the desmoid tumor (3,148). Cyclin-D1 is expressed in the nuclei in virtually all cases. These tumors are immunopositive for vimentin and CD34. Nuchal and Gardner fibromas have virtually identical pathologic features.
Palmar-plantar fibromatosis is seen in children but more commonly in adults. These are poorly circumscribed fibrous tumors with a pattern of spindle cell foci separated by bland hypocellular collagenized stroma (149).
Juvenile hyaline fibromatosis (JHF) is an autosomal recessive disorder, which is allelicallyrelated to infantile systemic hyalinosis (ISH) with a loss of function mutation in the capillary morphogenesis gene-2 (CMG2 on 4q21) (150,151). Large, painful nodules in the head and neck region (including marked gingival hypertrophy) and around joints evolve from small cutaneous papules, which are first noted in infancy and accelerate in growth throughout childhood. Osteolytic bone lesions develop, as well as joint contractures. Firm, white nodules in the soft tissues and dense fibrous effacement of the dermis, resembling to some extent morphea-scleroderma, are some of the pathologic features (Figure 25-35). The nodules are circumscribed and consist of homogeneously dense hyaline collagen with both paucicellular and hypercellular foci, consisting of ovoid stromal cells residing in apparent lacunae with a chondrocyte-like appearance (Figure 25-36). Unlike JHF, ISH has visceral involvement in addition to papulonodular lesions of the skin and soft tissues. The heart, intestinal tract, spleen, and skeletal muscle are infiltrated by the fibrohyaline tissue with a resemblance to amyloid. Protein-losing enteropathy can be a complication of small intestinal hyalinosis. Only infantile myofibromatosis among the other fibrous proliferations has visceral involvement by a more cellular, vasocentric nodular proliferation than the diffuse interstitial hyalinosis of ISH.
FIGURE 25-35 • JHF presented as a firm mass around the knee of a 17-year-old female who had several other similar masses excised previous to this one. This well-circumscribed mass had a glistening, slightly nodular appearance on cut surface. The consistency of the mass was described as firm with a chondroid-like quality.
FIGURE 25-36 • JHF presented as multiple masses in this 17-year-old female. The dense, hypocellular nodules of collagen are characteristic of this tumor. Inset: Rounded stromal cells within lacunar spaces resembling chondrocytes is another histologic feature. No other fibrous lesion in childhood approaches this degree of dense, uniform hyalinization with the possible exception of a Gardner fibroma which lacks nodularity.
FIGURE 25-37 • Calcifying aponeurotic fibroma in a 5-year-old male along the biceps tendon. A: Foci of dystrophic calcification within a nodular hyaline focus. B: Elsewhere the tumor had an infiltrative spindle cell pattern.
Calcifying aponeurotic fibromatosis (juvenile aponeurotic fibroma) is one of the least common of the fibrous tumors of childhood (1% to 2% of all cases) and has a predilection for the distal extremities (109). Usually, older children and adolescents present with a mass in the ankle or wrist in the deep subcutis, fascia, or tendon. A poorly circumscribed mass measuring less than 5 cm has a firm, gritty, gray-white appearance of cut surface. An infiltrative process of spindle cells (fibroblasts) is accompanied by less cellular, hyalinized areas in which foci of granular calcifications are found (Figure 25-37). Without the calcifications, there is a resemblance to infantile subcutaneous fibromatosis. Recurrences are reported in 50% or more of cases. Because of the distal, periarticular localization, monophasic SS may be briefly considered in the differential diagnosis.
FIGURE 25-38 • JNA in an 11-year-old male with a large skull-based mass. A: This field shows the presence of regularly distributed vascular spaces in a dense fibrous stroma with uniform cellularity. Inset: The stromal cells have indistinct cellular borders. B: β-Catenin immunostain shows intense nuclear positivity. Inset: There is nuclear positivity for androgen receptor as well.
Juvenile nasopharyngeal angiofibroma (JNA) is a fibrovascular tumor which is more fibrous than vascular in terms of histogenesis. Most cases present in pubertal-aged males with epistaxis (152). These tumors are seen in the setting of FAP, and like desmoid-type fibromatosis, the nuclei of JNA express β-catenin and androgen receptor (153) (Figure. 25-38). Another susceptibility gene in the development of JNA is GSTM1 (1p13.3) in a Brazilian study (154). A firm lobulated or pedunculated mass measuring 5 to 10 cm is the gross appearance. A uniform population of spindled to stellate fibroblasts lacks a fasciculated pattern, but appears more diffuse and is interrupted by evenly distributed thin-walled vascular spaces. Mast cells are commonly distributed within the background. These tumors have pushing rather than the infiltrating borders of desmoid-type fibromatosis, but despite this difference, JNAs are known to recur locally (155).
Fibromatosis colli is infrequently seen as a surgical specimen though it is one of the more common fibrous tumors of childhood. There is a predilection for the right neck and spontaneous regression occurs in more than 90% of cases (109,156). A firm, white lobulated fibrous mass measuring 1 to 3 cm typically arises in the lower one-third of the sternocleidomastoid muscle where it has infiltrative borders like the desmoid-type fibromatosis. However, fibromatosis colli is usually more cellular than desmoid-type fibromatosis and has a less collagenized stroma. There is some similarity to NF, which infrequently arises in skeletal muscle.
Calcifying fibrous pseudotumor (CFP) is a fibroinflammatory tumefaction which is recognized in children and adults alike and has a broad anatomic distribution with a predilection for the extremities in children (157,158). Virtually no anatomic site is immune from CFP (153). A well circumscribed, firm fibrous mass can measure in excess of 20 cm or as small as 2 to 3 cm. Dense collagen with scattered lymphoplasmacytic infiltrates and small calcific deposits, some with psammoma-like features, are the characteristic microscopic features. The spindle cells are immunoreactive for CD34, and nonreactive for smooth muscle actin and ALK, unlike the inflammatory myofibroblastic tumor (IMT) (159). A CFP of the neck in a 5-week-old girl was immunopositive for factor XIIIa, raising the question whether these tumors are dendritic cell (DC) in origin and possibly related to juvenile xanthogranuloma (JXG) (160).
TABLE 25-8 SITES OF INFLAMMATORY MYOFIBROBLASTIC TUMOR IN THE FIRST TWO DECADES OF LIFE
Sites
No.
Mean Age and Range
Sex (M/F)
Total (%)
Abdomen
61 (50)
Small Intestine
36
9 y (3 mo-17 y)
13/23
Omentum-mesentery
Liver
11
4 y (2 mo-9 y)
6/5
Stomach
5
9 y (6-14 y)
2/3
Retroperitoneum
5
7 y (4 mo-14 y)
3/2
Pancreas
2
12 y, 7 y
1/1
Spleen
2
16 y, 9 y
2/0
Genitourinary Tract
16 (13)
Bladder
13
11 y (4-17 y)
6/7
Scrotum
2
7 y, 15 y
2/0
Prostate
1
17 y
1/0
Thorax
32 (26)
Lung
17
10 y (2-20 y)
13/4
Larynx-Trachea
6
7 y (12 days-12 y)
3/3
Heart
9
2½ y (1 mo-12 y)
6/3
Superficial and Deep Soft Tissues
12 (10)
Extremities-trunk
5
6 y (1 mo-13 y)
3/2
Head and neck
3
2 y (1 y-3 y)
2/1
Perirectal-pelvic
4
8 y (3 y-13 y)
1/3
Brain
2 (2)
2
1 y, 15 y
0/2
123 (˜100)
From the files of the Lauren V. Ackerman Laboratory of Surgical Pathology, St. Louis Children’s Hospital, Washington University Medical Center, St. Louis, MO.
Inflammatory myofibroblastic tumor (IMT) is a distinctive clinicopathologic entity, which has emerged from the somewhat poorly defined group of idiopathic fibroinflammatory processes collectively known as inflammatory pseudotumors (161). In the WHO classification, the IMT is regarded as an “intermediate, rarely metastasizing” neoplasm, together with DFSP, SFT, low grade myofibroblastic sarcoma, myxoinflammatory fibroblastic sarcoma (MIFS) and CIF (5). IMT is typically seen in the first three decades with cases occurring as early as the first year of life and into early adulthood with a mean age at diagnosis between 10 and 15 years without the inclusion of older adults (3,109). The lung, gastrointestinal (GI) tract, mesentery, liver, and bladder are the principal primary sites, which in aggregate account for 70% to 75% of all cases in children (Table 25-8). This tumor is also ubiquitous in terms of its other less common sites of presentation including the dura, orbit, kidney, uterus, and upper respiratory tract. Peripheral soft tissues and bones are infrequently affected. In a small proportion of cases, multiple lesions may be detected at presentation or develop over the evolving clinical course. It is not always clear whether multiple IMTs are metastases or independently developing multifocal tumors (162). Constitutional or B-symptoms with fever, failure to thrive, and weight loss together with microcytic hypochromic anemia and polyclonal gammopathy are present in 5% to 15% of cases; these children may be a diagnostic dilemma for weeks to months. There is IL-6 production in association with IMTs, which often falls to normal levels after surgical resection.
FIGURE 25-39 • IMT in a 19-year-old male arising in the region of the inferior vena cava. The mass measures almost 7 cm in greatest dimension (51 g) and has a homogenous pale tan-white cut surface.
The tumors range in size from less than 1 to 15 cm in greatest dimension, with the larger IMTs arising in the abdomen. In the lung, IMT measures 4 to 6 cm, but in the mesentery or retroperitoneum, IMT is generally in excess of 10 cm. A well-circumscribed, non-encapsulated tumor has a glistening tan-white to gray-tan homogeneous and nodular appearance, with minimal hemorrhage and absence of necrosis in most cases (Figure 25-39). Calcifications are seen more often in the pulmonary IMTs (where it is one of the more common primary neoplasms of the lung in childhood) (163,164). Microscopically, three basic patterns are recognized; they are not necessarily in equal proportions nor is each represented in every tumor. The first of these is characterized by a dense spindle cell proliferation with the formation of fascicles and a variably prominent population of lymphocytes and mature plasma cells in the background. Adjacent foci may be composed of loosely arranged spindled to plump stromal cells in a myxoid and edematous background resembling NF to yet a third pattern of paucicellular dense fibrosis with some inflammatory cells in the background (Figure 25-40A-D). Dystrophic calcifications, osseous metaplasia, and collections of histiocytes are other features. Mitotic figures are found in the spindle cell foci; however, atypical mitotic figures and anaplasia should suggest a high-grade pleomorphic sarcoma. Necrosis is present in those IMTs which have undergone sarcomatous transformation and may be accompanied by overt nuclear pleomorphism and hyperchromatism. Those IMTs arising in the abdomen which are multifocal and are composed of polygonal or epithelioid cells tend to behave in an aggressive manner, more so than those tumors with the typical histologic features (161). Most IMTs are immunoreactive for vimentin and SMA (Figure 25-40C), as well as cytokeratin in a minority of cases in children. Approximately 50% to 60% of IMTs are ALK-1 positive with a membranous and/or cytoplasmic pattern reflecting a specific ALK-1 translocation (Figure 25-40D) (161,162) (Table 25-1). Coffin and associates (165) found that ALK-1-positive IMTs pursue a less aggressive course than those which are ALK-1 negative and may represent another tumor type. Those IMTs with a prominent epithelioid or round cell population are seemingly more aggressive (166). Attention has also been directed to the relationship, if any, between IMT with its often prominent population of polyclonal plasma cells and the IgG4-related sclerosing disorders (167). Surgical resection is the treatment of choice with a recurrence-free survival of 80% or greater. It is now recognized that there is an ALK-positive family of neoplasms beyond anaplastic large cell lymphoma and IMT (168). In a minority of IMTs, ROS-1 translocation has been reported (169).
FIGURE 25-40 • IMT in a 19-year-old male (see Figure 25-39). A: A uniform spindle cell proliferation is seen with few inflammatory cells (mainly plasma cells) in the background. B: The spindle cells are immunopositive for smooth muscle actin and also note the inflammatory cells in the background. C: Intense ALK-1 cytoplasmic positivity is shown.
The differential diagnosis includes low-grade myofibroblastic sarcoma, NF, inflammatory leiomyosarcoma, myxofibrosarcoma and CFP, desmoid-type fibromatosis, and gastrointestinal stromal tumor (GIST). The latter two neoplasms, when arising in the mesentery or intestine, display β-catenin (nuclear) and CD117 immunopositivity, respectively.
Myxoinflammatory fibroblastic sarcoma (MIFS) is an uncommon neoplasm with a predilection for the distal extremities mainly in adults, but is documented in older children and adolescents in nonacral sites (170,171). A sharply demarcated, lobulated or nodular tumor has a fibrous or fibromyxoid appearance. There is considerable microscopic variation in the stroma and spindled and epithelioid cells (Figure 25-41). These tumors are immunoreactive for D2-40, CD34, and CD68. It is thought that MIFS is one in a spectrum of related tumors including the hemosiderotic fibrolipomatous tumor and pleomorphic hyalinizing angiectatic tumor; these tumors appear to share a translocation involving rearrangements of TGFBR3 and/or MGEA5.
FIGURE 25-41 • MIFS presenting in the left axilla of a 10-year-old male. A: The mass is well circumscribed, measuring 6 × 5 × 4.5 cm, and has a glistening, firm tan surface. B: Spindle- and polygonal-shaped tumor cells are accompanied by lymphocytes, plasma cells, and a myxoid background. C: Ganglion-like cells and polygonal cells with vacuoles are seen. Inset: CD34 (shown), S100 protein, smooth muscle actin, and CD68 were expressed by the tumor cells.
Low-grade myofibroblastic sarcoma is a rare sarcoma with many overlapping morphologic and immunohistochemical features in common with myofibroma and IMT (172). There is a preference for the head and neck region in children (113). A superficial biopsy can be misleading especially given the resemblance to myofibroma or fibromatosis.
Fibrosarcoma (FS) with the exception of some specific histologic types is largely a diagnosis of exclusion and in children as such is uncommon (172). Monophasic SS, spindle cell RMS and malignant peripheralnerve sheath tumor are the three STS in children with FS-like spindle cell morphology.
Age range at diagnosis: 5 day-25 months (mean, 7.3 months; median, 5 months)
Sites:
Lower extremity
5
Trunk
5
Small intestine
3
Gluteal region
2
Intraabdominal
2
Upper extremity
1
Intrathoracic
1
Neck
1
Lung metastasis
2
aFrom the files (1989-2014) of the Lauren V. Ackerman Laboratory of Surgical Pathology, St. Louis Children’s Hospital, Washington University Medical Center, St. Louis, MO.
Congenital infantile fibrosarcoma (CIFS) is discussed in this section though it resides in the intermediate category of rarely metastasizing fibrous neoplasms. In general, there is a preference for the extremities (lower greater than upper) where the tumor presents either proximally or distally as a rapidly enlarging mass (173,174). Our own experience revealed 20 cases, virtually all presenting before 2 years of age, and 5 cases were diagnosed in the first month of life; the extremities including the gluteal region and trunk accounted for 60% of our cases (Table 25-9). We have also seen a few examples arising in the small or large intestine (Figure 25-42). These tumors can be quite hemorrhagic and may be mistaken for a vascular tumor clinically. Resected tumors are generally examined after preoperative chemotherapy which substantially alters the gross features including the size of the mass. A circumscribed, but non-encapsulated mass measuring 6 to 15 cm in greatest dimension has either a uniform, glistening tan-white cut surface or a cystic, hemorrhagic, and friable character in those cases without adjuvant therapy before surgery. Fascicles of uniform spindle cells with or without the so-called herringbone pattern are the classical features, but we have been impressed by the histologic diversity of these tumors with a poorly organized pattern of immature and even primitive mesenchymal cells. Whether in skeletal muscle or small intestine, the highly infiltrative character of the tumor can be appreciated (Figure 25-43A-D). The hemorrhagic appearance reflects the absence of a supporting stroma and the presence of pooled erythrocytes has a resemblance to peliosis. The individual tumor cells have elongated uniform nuclei, and mitotic figures are found without difficulty (Figure 25-43B). If there are an appreciable degree of pleomorphism and atypical mitotic figures, the tumor may represent something other than CIFs such as a spindle cell RMS. If the tumor is especially primitive with a myxoid background, ERMS, primitive myxoid mesenchymal tumor of infancy (PMMTI), MRT, and undifferentiated round cell sarcoma with Ewing-like features are other considerations in the differential diagnosis in a child less than 2 years old. CIFS has the ETV6-NTRK3 fusion transcript, t(12;15)(p13;q25), in some 70% of cases (173,174,175). If the translocation is not identified, but the tumor otherwise satisfies the morphologic features then a diagnosis of CIFS should be made (175). These tumors have an immunophenotype restricted to vimentin positivity (Figure 25-43D). Foci resembling CIFS may be found in other fibrous tumors of childhood including infantile myofibroma, but the characteristic translocation is not present in these cases (176,177). The other neoplasms with the t(12;15) translocation are cellular mesoblastic nephroma or infantile FS of the kidney, juvenile or secretory carcinoma of the breast and salivary gland. After chemotherapy, the resected specimen may have minimal residual tumor with only fibrosis, histiocytes, and hemosiderin deposition. Metastasis occurs in less than 5% of cases. In the lung, the infantile peribronchial myofibroblastic tumor has a histologic resemblance to CIFS but lacks the signature translocation of the latter tumor (178). Primitive myxoid mesenchymal tumor of infancy is represented by less than 15 cases in the literature of a soft tissue neoplasm on the trunk or extremities mainly in infants to the age of 15 months (179). These soft, often gelatinous tumors are composed of uniform spindle cells in a myxoid matrix except at the periphery where the tumor is more cellular and has a collagenized stroma (180). There is some resemblance to CIF, but with an absence of the t(12;15) translocation. PMMTI should be regarded as a low grade malignant neoplasm with the potential to progress to a high grade sarcoma (181).
FIGURE 25-42 • CIFS of the jejunum in a 10-day-old male who presented with intestinal obstruction. A: A uniform spindle cell proliferation involves the entire thickness of the bowel wall. B: The spindle cells infiltrate into the lamina propria.
FIGURE 25-43 • CIFS presenting in the soft tissues of the posterior trunk in a 4 month-old female. A: The spindle cell pattern forming short fascicles presents the differential diagnosis of spindle cell rhabdomyosarcoma. Focal areas of hemorrhage resulted in the clinical impression of a hemangioma. B: The infiltrative character of the tumor is seen in this focus of residual adipose tissue in the background. Inset: All muscle markers were nonreactive with only immunopositivity for vimentin.
FIGURE 25-44 • LGFS of the lower extremity in a 10-year-old male. A: The tumor measuring 3.5 cm in greatest dimension was well circumscribed with a delicate capsule as seen in this low-magnification field. B: An alternating myxoid and more cellular focus.
Low-grade fibromyxoid sarcoma (LGFS, Evans tumor) is a generally slow-growing soft-tissue neoplasm with a preference for the lower extremity and trunk but has a wide anatomic distribution (182,183). Approximately 20% of cases are discovered before the age of 20 years and have been seen as early as 4 years of age (184). A well-circumscribed, fibrous-appearing tumor with a sharply demarcated pseudocapsule has a distinctive microscopic appearance of bland spindle cells with an alternating pale myxoid background to a more collagenous stroma (Figure 25-44A-D). Foci of epithelioid cells are found in those tumors with a hyalinized stroma and resemble the related sclerosing epithelioid fibrosarcoma (SEF) and in some of these cases, hyalinizing giant rosettes are present to establish the linkage among LGFS, hyalinizing spindle cell tumor with giant rosettes and the SEF (Figure 25-45). Attenuated, uniform spindle cells with fibroblast-like features have low grade nuclear features and mitotic figures are found only with patience. Perivascular hypercellularity is a consistent finding. A perineural nerve sheath tumor, fibromatosis, SFT, DFSP with fibromyxoid features, or perineurioma are the differential diagnostic considerations. Until recently, LGFMSs were largely restricted to vimentin positivity, but the upregulation of the mucin 4 gene (MUC4) has proven to be a sensitive marker, as well as for SEF, not surprisingly (185) (Figure 25-44D). Two translocations with FUS and CREBBL2 (90% of cases) or CREB3L1 in the fusion transcripts, t(7;16) (q32-34;P11) or t(11;16) (p11;p11) can be demonstrated as FUS breakaparts by FISH. A more recent observation is the identification of a EWSR1-CREB3L1 fusion transcript in LGFMS (186).
Only gold members can continue reading. Log In or Register to continue