Soft tissue tumors and tumor-like conditions, a highly heterogeneous group of lesions, are classified on the basis of their overall microscopic appearance, tissue differentiation pattern, clinical presentation, and biologic potential. In general, they are classified into several major histogenetic categories according to their exclusive or dominant lineage differentiation pattern. Within each major histogenetic category, the lesions are further separated into benign and malignant tumors. Benign tumors have limited local growth potential. They typically grow in a noninvasive expansile fashion and in the vast majority of cases can be eradicated by conservative excision. In contrast, malignant soft tissue tumors or sarcomas are at least locally aggressive and grow in an invasive, destructive fashion. Clinical behavior of these tumors is characterized by continuous growth with multiple local recurrences and, sometimes, distant metastasis. The clinical aggressiveness varies among different categories of malignant tumors and even among individual tumors of the same class. As a group, sarcomas are characterized by a relatively high incidence of distant blood-borne metastases primarily to the lungs, but some of them may initially metastasize to regional lymph nodes.
Soft-tissue sarcomas are less common than epithelial malignancies and constitute less than 1% of human cancers. Over the 15-year period of 1973-1987, 19,684 sarcomas were reported to the National Cancer Institute’s Surveillance Epidemiology and End Results Program, and more than half of them (10,034 cases) arose in soft tissue. The incidence rates calculated on the basis of these data show gradual increases in relation to age. The overall age-adjusted incidence rate is 2.4 and 1.6 per 100,000-individuals for males and females respectively. Malignant fibrous histiocytoma (20% of all soft tissue sarcomas) is the most frequently diagnosed sarcoma followed by liposarcoma (18.3%), leiomyosarcoma (8.8%), fibrosarcoma (8.0%), rhabdomyosarcoma (6.7%), Kaposi’s sarcoma (3.4%), synovial sarcoma (3.9%), and malignant peripheral nerve sheath tumor (3.6%). Median age at diagnosis for all soft tissue sarcomas is 54.8 years for males and 55.3 years for females.
The clinical course of sarcomas varies not only among tumors of different types but also among individual lesions of the same type. Therefore, in addition to histologic type, such information as grade and stage are important in assessing prognosis. The original grading system proposed by Broders (1922, 1939) was based on microscopic features such as (1) cellularity, (2) pleomorphism, (3) mitotic activity, (4) necrosis, (5) type of growth (expansive versus invasive). In reference to sarcomas two of their microscopic features, i.e., the frequency of mitotic figures and the extent of necrosis, are the most important parameters for histologic grading. The most frequently used three-tier system separates tumors into a well-differentiated category (grade 1) and a poorly differentiated category (grade 3). The intermediate tumors are classified as grade 2 lesions. The frequently used four-tier system shows minimal differences between the two lower grades (grades 1 and 2) and two higher grades (grades 3 and 4). For practical use, the soft tissue sarcomas are frequently divided into two major categories, low and high grade. The low-grade category typically signifies a predominantly locally aggressive tumor with minimal or no potential for metastases. The lesions designated as high-grade tumors typically have a high propensity for distant metastasis.
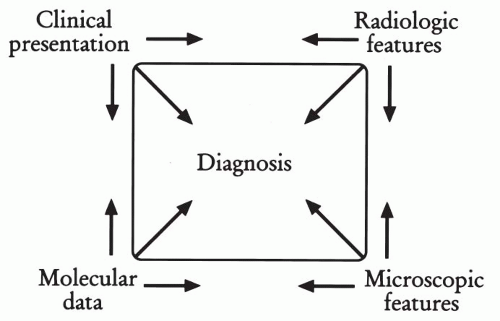
Figure 35-1 Analytical approach to diagnosis of soft tissue tumors. The step-wise approach in which clinical, radiographic, microscopic, and molecular data are taken into consideration in establishing the diagnosis. (Adapted from Dorfman H, Czerniak B. Bone Tumors, St. Louis, Mosby, 1998.)
In this chapter we retain the conventional approach to the classification of soft tissue sarcomas, dividing them into several major histogenetic categories based on their overall microscopic appearance, tissue differentiation pattern, and biologic potential. We advocate a multimodal approach, graphically depicted as the so-called diagnostic quadrangle, which describes a step-wise analysis in which four distinctive data sets—clinical, radiographic, microscopic, and molecular—are considered to establish the diagnosis and treatment plan (Fig. 35-1). Such step-wise analysis is more likely to lead to consistency and accuracy than an intuitive approach based on fragmentary data.
BIOPSY TECHNIQUES OF SOFT TISSUE LESIONS
Planning biopsies of soft tissue lesions is a complex procedure, as it must take into account the technical aspects of definitive surgery. Inappropriately performed biopsies can complicate subsequent surgery or even eliminate some treatment options. For example, an incorrectly placed biopsy incision may make optimal limb-sparing procedures impossible because of the danger of tumor seeding at planned excision margins. The ideal approach begins with preoperative consultations among the surgeon, medical oncologist, radiologist, and pathologist to understand the clinical setting and to establish the optimal diagnostic and therapeutic plan.
Frequently, needle aspiration cytology, under the guidance of radiographic imaging techniques, is used as a preliminary diagnostic approach that is particularly valuable when the lesion is not readily accessible to open biopsy. Unfortunately, in many instances, the aspirated samples do not provide sufficient material for final diagnosis. The lesions that are extremely difficult or even impossible to diagnose from limited material provided by fine needle aspirations or core needle biopsies require open incisional or excisional biopsy.
Cytological preparations such as touch smears, scrapes, or needle aspirations can assist in the intraoperative assessment of open biopsy material and can provide valuable information on the morphology of individual cells and the architecture of cell clusters not distorted by freezing artifacts. At the time of preliminary examination of material obtained by closed or open biopsy technique, a decision must be made as to whether the specimen requires some additional diagnostic procedures such as immunochemistry electron microscopy, cytogenetics, or other molecular analysis.
FUNDAMENTALS OF MOLECULAR BIOLOGY OF SOFT TISSUE TUMORS
During the last two decades our knowledge of the molecular events responsible for the development and progression of human tumors, including those that arise in the soft tissue, has dramatically increased because of numerous technological developments (Busam and Fletcher, 1997). The application of these techniques to the study of soft tissue tumors has led to a significant increase in new information about the molecular mechanisms responsible for malignant transformation of these rare, poorly understood neoplasms (Anderson et al, 1999; Argatoff et al, 1996).
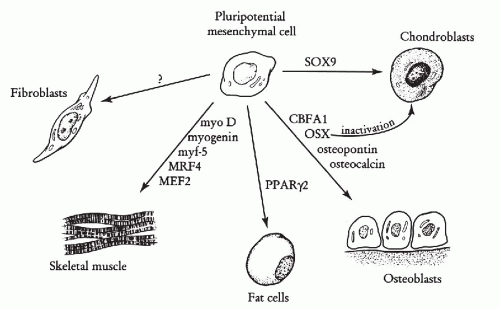
The molecular regulation of mesenchymal cell differentiation is only superficially known. All of the five major lineages of mesenchymal origin (chondroblastic, osteoblastic, lipoblastic, muscular, and fibroblastic) originate from pluripotential mesenchymal cells. Some of the major master genes responsible for triggering lineage differentiation for pluripotential mesenchymal cells are shown in Figure 35-2. These genes trigger the activation of lineage-specific genes responsible for unique phenotypic change of responsive cells (Rodan and Harada, 1997). The myoD gene serves as a paradigm of such master regulators, causing expression of genes characteristic of the muscle phenotype.
Unique phenotypic features of distinct mesenchymal lineages and their tumors aid in the classification of soft tissue neoplasms. Unfortunately, the tumors that originate from individual lineages do not always exhibit the same phenotype. A good example of such an abnormal phenotype is a rare expression of cytokeratins in Ewing’s sarcoma/PNET or characteristic coexpression of desmin and cytokeratins in small cell desmoplastic tumor. Therefore, the classification of the tumor on the basis of its phenotypic features by immunohistochemistry is complex and requires knowledge not only of normal phenotype of each individual mesenchymal lineage but also of a large array of aberrant features found in individual tumor types. The purpose of this summary is to present the diagnostic impact of various molecular features on the differential diagnosis of soft tissue tumors.
Figure 35-2 Master genes responsible for lineage differentiation for pluripotential mesenchymal cells. MyoD gene causes myogenic differentiation by induction of myogenin, myf-5, MRF4, and MEF2 genes. PPAR γ2 (peroxisome proliferation activator receptor γ2) is responsible for adipocyte differentiation. CBFA1 and OSX by induction of osteopontin and osteocalcin genes are master regulators for osteoblastic differentiation. Sox9 is responsible for chondroblastic differentiation and is up-regulated in prechondroblastic mesenchymal condensation. Moreover, inactivation of OSX diverge preosteoblastic cells towards chondroblastic lineage. (Modified from Rodan GA, Harada SI. The missing bone. Cell 89:677-680, 1997).
Many soft tissue sarcomas exhibit pronounced aneuploidy with alterations of multiple chromosomes. However, in contrast to more common epithelial neoplasms, the soft tissue tumors have a high incidence of specific chromosomal translocations associated with formation of novel tumor-specific chimeric genes (Fletcher et al, 1991a; May et al, 1993; Sandberg and Bridge, 1994; Sreekantaiah et al, 1994; Ladanyi, 1995). The specificity of these translocations and their gene products is becoming an integral part of the diagnostic work-up of some soft tissue neoplasms. These features are especially helpful in cases that are histologically, pathologically, and clinically equivocal (Ladanyi and Gerals, 1994; Panagopoulos et al, 2002; Sandberg and Bridge, 2002). Diagnostically valid chromosomal translocations and their associated chimeric genes are summarized in Table 35-1. More detailed discussion of these translocations is provided under sections devoted to specific tumors.
TABLE 35-1 CHROMOSOMAL TRANSLOCATION AND GENE REARRANGEMENTS IN SOFT TISSUE SARCOMAS
Tumor Type
Cytogenetics
Genes Involved
EWS/PNET
t(11;22) (q24;q12)
FLI-1-EWS
t(21;22) (q22;q12)
ERG-EWS
t(7;22) (p22;q12)
ETV1-EWS
Desmoplastic small round cell tumor
t(11;22) (q13;q12)
WT1-EWS
Extraskeletal myxoid chondrosarcoma
t(9;22) (q22;q12)
CHN-EWS
t(9;17) (q22;q11)
TEC-TAF2N
t(9;15) (q22;q21)
TEC-TCF12
Clear cell sarcoma
t(12;22) (q13;q12)
ATF-1-EWS
Alveolar rhabdomyosarcoma
t(2;13) (q35;q14)
PAX3-FKHR
t(1;13) (p36;q14)
PAX7-FKHR
Myxoid and round cell liposarcoma
t(12;16) (q13;p11)
CHOP-TLS
t(12;22) (q13;q11)
CHOP-EWS
Synovial sarcoma
t(x;18) (p11;q11)
SSX1-SYT
SSX2-SYT
Although chromosomal translocation and the resulting chimeric genes represent prototypic diagnostically valid molecular abnormalities found in soft tissue tumors, other less specific genetic changes, such as chromosomal deletions or gains, can also be diagnostically useful. A selected list of various genetic changes other than translocations found in some benign and malignant soft tissue tumors is provided in Table 35-2.
TABLE 35-2 CHROMOSOMAL ABNORMALITIES OTHER THAN TRANSLOCATION IN BENIGN AND MALIGNANT SOFT TISSUE TUMORS
The lesions of fibrohistiocytic tissue can be divided into three major groups: (1) clinically benign proliferations that typically do not recur after conservative local excision, (2) ill-defined local proliferations with high propensity for local recurrence but virtually no metastatic potential, (3) aggressive, fully malignant tumors with high propensity for distant metastasis.
Benign Lesions
Fibrous Histiocytoma
Pathology and Histology
Fibrous histiocytoma is a frequent, benign, slowly growing cutaneous lesion of adults with predilection for extremities (Gonzalez and Duarte, 1982; Fletcher, 1990). Less frequently it presents as a subcutaneous lesion without skin involvement. It is extremely rare in deep soft tissue, parenchymal organs, or bone (Meister et al, 1978b). The vast majority of lesions are not larger than 2 cm in diameter. They usually present as solitary flat skin lesions, but in 30% of the cases they are multifocal. Approximately 5% to 10% of the lesions recur after simple excision. Fibrous histiocytoma grows as a relatively well circumscribed nodule composed of a mixture of spindle and plump oval cells arranged in ill-defined fascicles forming a criss-cross pattern. Foamy histiocytic cells and hemosiderin deposits as well as multinucleated giant cells with peripheral nuclei of Touton type can be present. The lesion is typically separated from the overlying epidermis, but its deep aspects may show irregular infiltration of the subcutis.
Immunohistochemically fibrous histiocytomas express factor XIIIa and tinascin, which suggest that these tumors arise from dermal dendrocytes (Reid et al, 1986; Cerio et al, 1990). Lack of CD34 expression is helpful in distinguishing fibrous histiocytoma from dermatofibrosarcoma protuberans, which is consistently positive for this marker (Kamino and Salcedo, 1999).
Cytology
Aspirates from fibrous histiocytoma are cellular and contain multiple clusters of tightly packed cells. They represent a mixture of plump spindle, oval, or polygonal cells with vesicular nuclei and small inconspicuous nucleoli. Rare multinucleated giant cells corresponding to Touton cells and foamy macrophages can be also found. Usually there is an admixture of inflammatory cells such as lymphocytes and plasma cells.
Fibromatoses
Pathology and Histology
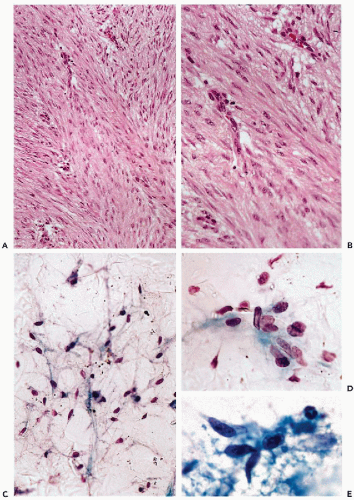
Fibromatoses comprise a wide spectrum of borderline fibrous tissue lesions involving various anatomic sites (Stout, 1961; Allen, 1977). The unifying theme is a multinodular diffuse proliferation of well-differentiated fibrous tissue with high propensity for local recurrence but virtually no metastatic potential (Fig. 35-3A,B). According to their anatomic location, fibromatoses are divided into two major groups: superficial and deep (Burke et al, 1990). Palmar fibromatosis is by far the most frequent superficial form of fibromatosis. Originally described by Dupuytren, it is still referred to as Dupuytren’s contracture (Gabbiani and Majno, 1972). Other typical sites of involvement by superficial fibromatoses are the hand (palmar fibromatosis), foot (plantar fibromatosis), and penis (penile fibromatosis). Congenital fibromatoses and fibromatoses of infancy often affect the head and neck area.
Deep soft tissue musculo-aponeurotic fibromatoses have predilection for the shoulder, chest wall, and thigh (Enzinger and Shiraki, 1967). Desmoid tumor involving the abdominal muscular structures of young women during or following pregnancy is a prototypic fibromatosis related to trauma (Karakousis et al, 1993). A distinct group of intraabdominal mesenteric fibromatoses is associated with Gardner’s syndrome (Naylor et al, 1979). Idiopathic retroperitoneal fibrosis is discussed in Chapter 40.
Cells of fibromatosis express vimentin, and different muscle markers such as smooth muscle actin, muscle-specific actin, and desmin. Expression of these markers is variable and depends on the degree of myofibroblastic differentiation. Expression of CD117 (c-kit), originally considered to be characteristic of gastrointestinal stromal tumor (GIST), is present in nearly 75% of the cases. In contrast to GIST, fibromatoses are consistently negative for CD34 (see Chap. 24).
Cytology
Superficial fibromatoses are practically never diagnosed by cytology. Fine-needle aspirations of deep lesions are performed in order to rule out high-grade sarcomas. The aspirates contain large, three-dimensional fragments of fibrous tissue and sparse isolated fibroblast-like spindle cells (Sauer et al, 1997; Pereira et al, 1999; Kurtycz et al, 2000). There is no cytologic atypia or mitotic activity (Fig. 35-3C-E) (Liu et al, 1999a).
Malignant Lesions
Dermatofibrosarcoma Protuberans
Pathology and Histology
Dermatofibrosarcoma protuberans, a tumor of intermediate grade of malignancy, typically presents as a cutaneous mass with predilection for the trunk and proximal portions of the extremities (Fletcher et al, 1985). It primarily affects individuals during their early or middle adult life. Early lesions present as flat indurations of the skin, enlarging to produce a multinodular, protruding mass (Burkhardt et al, 1966). The tumor is a locally aggressive lesion with high propensity for local recurrence (approximately 50%) and only minimal metastatic potential (Burla and Gotz, 1965; Connelly and Evans, 1992; Mentzel et al, 1998). Microscopically it is composed of densely packed short spindle cells arranged in a distinct storiform pattern that show only minimal nuclear atypia. Tumor diffusely infiltrates the dermis and subcutis, creating a very characteristic lace-like pattern at its periphery (Fig. 35-4A,B). In rare instances dermatofibrosarcoma protuberans can progress to a low-grade or even high-grade fibrosarcoma with malignant fibrous histiocytoma-like features (Mentzel et al, 1998). Surprisingly, several recent studies indicate that this progression does not significantly increase the local recurrence rate or metastatic potential.
Immunohistochemically, dermatofibrosarcoma is characterized by expression of CD34, suggesting its origin from CD34-positive dermal dendritic cells (Goldblum and Tuthill, 1997) Cytogenetically, dermatofibrosarcoma protuberans is characterized by supernumerary chromosomes consisting of amplified sequences from chromosomes 17 and 22. Fusion of the PDGFβ-chain gene with COL1A1 (collagen 1α1 gene) puts the PDGFβ gene under control of the COL1A1 promoter, creating a novel fusion protein.
Figure 35-3 Fibromatosis.A,B. Histologic appearance of fibromatosis. Note uniform proliferation of fibroblastic cells showing no atypia and only minimal mitotic activity. C-E. Cytologic features of fibromatosis. Note isolated spindly fibroblastic cells with indistinct cytoplasm. Benign looking nuclei with evenly distributed chromatin show neither hyperchromasia nor nucleoli.
Cytology
Cytologic preparations from dermatofibrosarcoma protuberans are highly cellular and consist mostly of large 3-dimensional aggregates of densely packed short spindle cells with plump ovoid nuclei and scant cytoplasm (Fig. 35-4C). Atypia is minimal and the nuclei contain evenly distributed chromatin with small, inconspicuous nucleoli (Filipowicz et al, 1999). Large cellular aggregates may contain prominent, branching capillary vessels surrounded by tightly packed tumor cells (Fig. 35-4D).
Fibrosarcoma
Pathology and Histology
The most significant difference between fibromatosis and fibrosarcoma is that the latter is capable of metastases (Stout, 1954; Oshiro et al, 1994). Fibrosarcoma is most frequent between ages 30 and 50 years and is predominantly found in deep soft tissue of the lower extremities (Scott et al, 1989). The characteristic microscopic feature is uniform growth of fibroblastic spindle cells with scanty cytoplasm separated by collagen deposits forming intersecting bundles reminiscent of herringbone (Fig. 35-5A,B). Tumors with admixture of inflammatory cells (inflammatory fibrosarcoma), tumors with stromal sclerosis and epithelioid change (sclerosing epithelioid sarcoma), and stromal myxoid change (low-grade fibromyxoid sarcoma) represent distinct clinicopathologic forms of fibrosarcoma (Evans, 1987; Meis-Kindblom et al, 1995; Dvornik et al, 1997; Eyden et al, 1998). Fibrosarcoma may also affect newborns and infants (congenital or infantile fibrosarcoma), in these cases typically arising in the extremities. Histologically they are similar to adult fibrosarcoma but are less aggressive (Schofield et al, 1994).
Figure 35-4 Dermatofibrosarcoma protuberans (DFSP).A,B. Histologic appearance of dermatofibrosarcoma protuberans. Neoplastic cells showing minimal atypia are arranged in storiform pattern. Note infiltration of tumor cells among adipocytes of subcutaneous fat. C,D. Cytologic features of dermatofibrosarcoma protuberans. Note large, 3-dimensional tissue fragments with discernable storiform pattern. The cells have scanty basophilic, elongated cytoplasms. The nuclei show evenly distributed chromatin and only small, inconspicuous nucleoli. Accumulations of cells around capillary vessels can be seen.
Immunohistochemically, fibrosarcoma express vimentin and may show scattered positivity for muscle markers (smooth muscle or muscle-specific actin) consistent with myofibroblastic differentiation. Negativity for epithelial markers is very useful in differential diagnosis with monophasic synovial sarcoma. Some special variants of fibrosarcoma such as sclerosing epithelioid type may, however, express epithelial membrane antigen and some neural markers including S-100 protein and neuron-specific enolase.
Cytology
Aspirations from fibrosarcoma are highly cellular and consist of loose clusters of spindle cells as well as many isolated single cells with elongated scanty cytoplasm and plump oval nuclei. The atypia is low to moderate, and the nuclei contain evenly distributed chromatin and small nucleoli (Fig. 35-5C,D). A fascicular arrangement of spindle cells is occasionally seen in larger clusters (Logrono et al, 1999a). Overall, cytologic features overlap those of other spindle cell tumors such as leiomyosarcoma, synovial sarcoma, and peripheral nerve sheath tumor (Liu et al, 1999b).
Malignant Fibrous Histiocytoma
Pathology and Histology
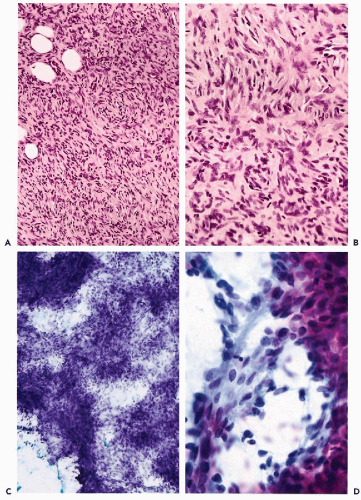
Malignant fibrous histiocytoma represents the most common soft tissue sarcoma of late adult life (Bertoni et al, 1985; Rooser et al, 1991; Fletcher, 1992). Tumors that belong to this category manifest a wide range of histologic appearances and can be subclassified into storiform-pleomorphic, myxoid, giant cell, and inflammatory subtypes (Weiss and Enzinger, 1978; Enzinger, 1986; Khalidi et al, 1997). The most frequent, the storiform-pleomorphic type, is characterized by a mixture of highly atypical spindle and pleomorphic cells arranged focally in a storiform pattern. There is usually an admixture of haphazardly scattered, highly atypical, multinucleated giant cells. Brisk mitotic activity with numerous atypical mitoses as well as areas of necrosis complement an overall highly malignant microscopic appearance of the tumor (Fig. 35-6A,B). Myxoid subtype is the second most frequent variant of malignant fibrous histiocytoma, characterized by prominent myxoid change in the stroma (Fig. 35-7A,B) (Weiss and Enzinger, 1977; Mentzel et al, 1996). Tumors with a diffuse prominent myxoid change behave less aggressively than do other types of malignant fibrous histiocytoma. Giant cell variant contains numerous osteoclast-like giant cells, while a significant admixture of inflammatory cells is seen in a subset of these tumors referred to as inflammatory malignant fibrous histiocytomas (Khalidi et al, 1997).
Immunohistochemistry has limited application in diagnosis of malignant fibrous histiocytoma. The main role for special stains is to rule out specific lineage of differentiation such as epithelial, rhabdoid, or melanocytic.
Cytology
Aspirates from malignant fibrous histiocytoma are highly cellular and contain obviously malignant, highly pleomorphic, cancer cells with eosinophilic cytoplasm arranged in large sheets, and small clusters or as individual cells. The cells vary in size and shape. The nuclei of the tumor cells show pronounced atypia with irregular clumped chromatin granules and prominent nucleoli (Fig. 35-6C,D). Multinucleated giant cells as well as cells with bizarre nuclei are common. Cellular debris reflecting tumor necrosis and an admixture of inflammatory cells can be present. Tumor cells show brisk mitotic activity—atypical mitoses are easy to find (Berardo et al, 1997
Only gold members can continue reading. Log In or Register to continue