decades has indicated that the success of any immunohistochemical procedure depends on multiple factors, such as the antigen preservation, the specificity and avidity of the primary antibody and the sensitivity of the detection system. These factors are interrelated; reliable and reproducible results can be achieved only when each variable is optimized.
may result in a mixture of formalin fixation and ethanol fixation, the latter resulting from tissue processing in graded ethanols. Brief formalin fixation will result in cross-link formation only at the periphery of the block while the center will be fixed in ethanol. As a result, sections prepared from such blocks may show variable staining that is more or less intense staining at the center or periphery, depending on the reactivity of the antibody and the retrieval procedure employed (Werner et al, 2000).
TABLE 45-1 IMMUNOCYTOCHEMISTRY USING DIFFERENT TYPES OF CYTOLOGICAL PREPARATIONS | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
precipitating or coagulating fixatives rather than cross-linking reagents; accordingly, retrieval procedures are not generally useful in restoring reactivity following fixation with these reagents. However, for some antigens a retrieval step may help in such preparations provided that the cells have been collected on adhesive slides.
of DNA and RNA from archival samples. Heat induced antigen retrieval may also result in enhanced reactivity of endogenous biotin. This is particularly problematic in mitochondria-rich cells. When using an avidin biotin-based detection system, therefore, it is critical that endogenous biotin be blocked with avidin prior to staining (Rodriquez-Soto et al, 1997) or that a non-avidin—biotin system be used.
glucose oxidase) can be used in the antibody labeling systems (Lam et al, 1988; Gown, 1988).
TABLE 45-2 FALSE-POSITIVE STAINING | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
specimen and control are subjected simultaneously to identical fixation, processing, retrieval, staining, and interpretation. This approach, however, does not circumvent other important preanalytic variables, such as antigen degradation during transport of fresh specimens or fixation time if the specimens are delivered to the laboratory in the fixed state.
TABLE 45-3 INTERMEDIATE FILAMENTS | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
that react with keratins 1 through 8 and 14 through 19.
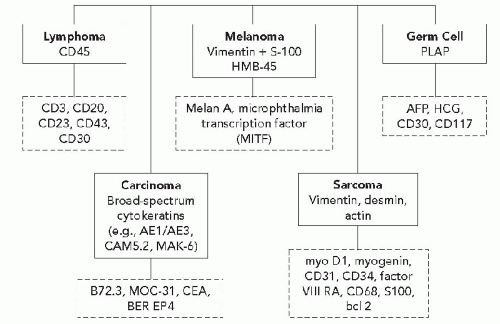 Diagram 45-1 Immunochemical approach to the classification of undifferentiated tumors. Closed boxes indicate recommended antibodies for primary screening of cases. Dashed boxes indicate antibodies for additional characterization of these tumors. |
and Ewing’s tumor. Melanomas, particularly in metastatic sites, may also be positive for cytokeratins and occasional examples of malignant lymphoma, particularly of the Ki-1 and large cell types, have been reported to be positive for cytokeratins. Immunoreactivity for cytokeratins (AE1/AE3) also occurs in glioblastoma multiforme and this finding has been attributed to cross reactivity with epitopes present in the neoplastic glial cells (Morrison and Prayson, 2000). This is particularly problematic when the differential diagnosis includes metastatic poorly differentiated carcinoma. Oh and Prayson (1999) have shown that more than 50% of cases of glioblastoma multiforme are reactive for AE1/AE3 while only 1 of 23 cases contained cells that were positive for CAM5.2 and cytokeratins 7 and 20. In contrast, metastatic carcinomas were positive both for AE1/AE3 and CAM5.2.
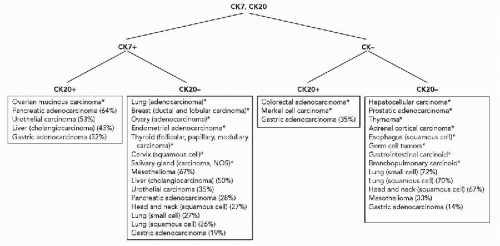 Diagram 45-2 Differential expression of cytokeratins 7 and 20. *More than 75% of cases are positive or negative. (Data from Wang et al, 1995; Blumenfeld et al, 1999; Tot, 1999; Chu et al, 2000A.) |
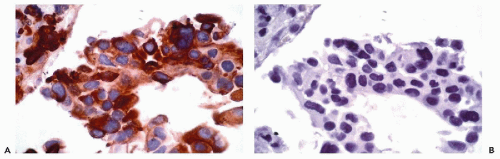 Figure 45-1 Poorly differentiated adenocarcinoma of the lung. A. Immunoperoxidase stain for cytokeratin 7 in cell block demonstrates uniform positivity within the tumor cells. B. Immunoperoxidase stain for cytokeratin 20 is negative. |
for EMA. Perineural cells are also reactive for EMA and positive reactions for this marker are present in nerve sheath tumors. Other tumors that are positive for EMA include meningiomas and certain sarcomas (epithelioid sarcoma, synovial sarcoma, chordoma, and chondrosarcoma) (Fig. 45-4).
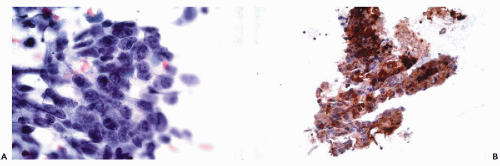 Figure 45-2 Metastatic colonic adenocarcinoma in liver. A. Fine needle aspiration biopsy stained with the Papanicolaou stain. B. Immunoperoxidase stain for cytokeratin 20 in cell block demonstrates uniform positivity within the tumor cells. |
and stimulate or suppress gene expression. Transcription factors may be tissue specific or may be present in a variety of different tissue types. Thyroid transcription factor-1 (TTF1), for example, is present in thyroid follicular cells and C-cells and is also present in the lung (Ordonez, 2000) (Fig. 45-7). The adrenal 4 site/steroidogenic factor is present in steroid-producing cells and in certain anterior pituitary cell types. The pituitary transcription factor, Pit-1, is present in certain cells of the anterior pituitary and is also present in the placenta (Kulig and Lloyd, 1996). In some instances, antibodies to transcription factors are of considerable value in determining the origins of tumors of unknown primary sites.
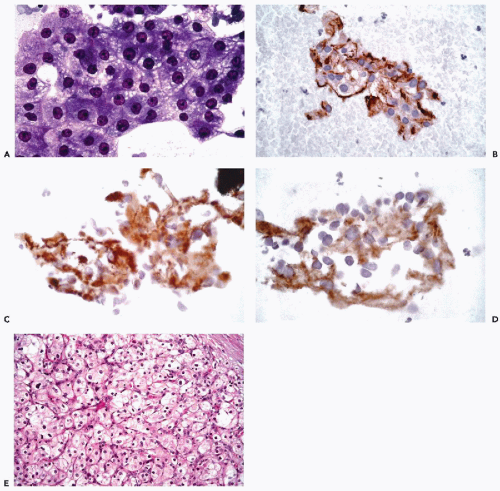 Figure 45-3 Metastatic renal cell carcinoma involving the lung. A. Fine needle aspiration biopsy stained with the Papanicolaou stain. B. Immunoperoxidase stain for EMA performed on destained slide demonstrates membrane positivity. C. Immunoperoxidase stain for cytokeratins 8 and 18 (CAM5.2) on a de-stained slide demonstrates cytoplasmic positivity. D. Immunoperoxidase stain for vimentin on a destained slide demonstrates focal cytoplasmic positivity. E. Histological section stained with hematoxylin and eosin demonstrates the typical features of renal cell carcinoma. |
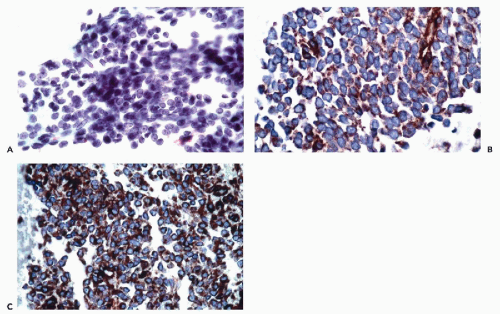 Figure 45-4 Metastatic synovial sarcoma involving the lung. A. Fine needle aspiration biopsy stained with the Papanicolaou stain. B. Immunoperoxidase stain for EMA in cell block demonstrate plasma membrane positivity. C. Immunoperoxidase stain for vimentin in cell block demonstrates diffuse cytoplasmic positivity. |
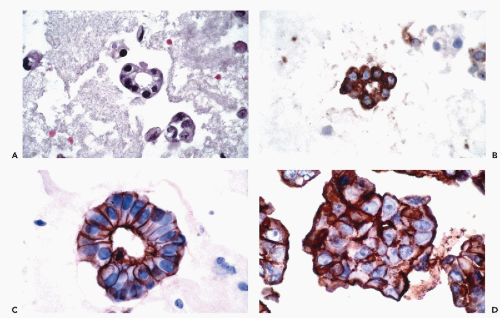 Figure 45-5 Metastatic adenocarcinoma in pleural effusion. A. Hematoxylin and eosin stain of cell block. B. Immunoperoxidase stain for CEA in cell block reveals cytoplasmic and membrane positivity. C,D. Immunoperoxidase stain for B72.3 also demonstrates plasma membrane positivity. |
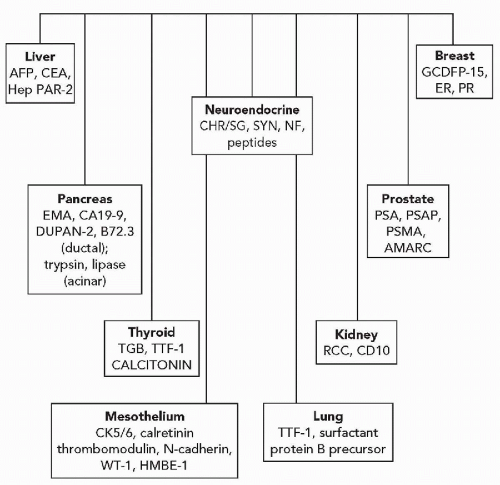 Diagram 45-3 Immunocytochemistry of poorly differentiated carcinomas. AFP, alpha fetoprotein; AMARC, alpha-methylacyl-coAracemase; CEA, carcinoembryonic antigen; CHR/SG, chromogranins/secretogranins; EMA, epithelial membrane antigen; ER, estrogen receptor; GCDFP-15, gross cystic disease fluid protein-15; HepPar 1, hepatocyte paratin antibody; NF, neurofilaments; PSA, prostate-specific antigen; PSMA, prostate-specific membrane antigen; PSAP, prostate-specific acid phosphatase; PR, progesterone receptor; RCC, renal cell carcinoma; SYN, synaptophysin; TGB, thyroglobulin; TTF1, thyroid transcription factor; WT-1, Wilms’ tumor gene protein. |
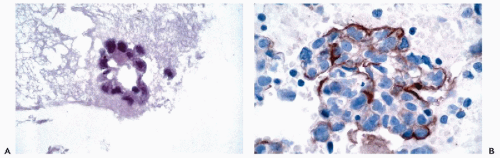 Figure 45-6 Ovarian papillary serous carcinoma involving the omentum. A. Hematoxylin and eosin stain of cell block. B. Immunoperoxidase stain for CA-125 in cell block demonstrates plasma membrane positivity. |
actin (HHF35), skeletal muscle or sarcomeric actin, myoglobin, and creatine phosphokinase-MM (Tsukada et al, 1987; Swanson and Wick, 1995; Suster, 2000) (Fig. 45-8). Desmin (MW-55,000) is the major intermediate filament type of muscle cells including cardiac, skeletal and smooth muscle cell types. Both desmin and muscle-specific actin are the most commonly used markers for muscle tumors (Truong et al, 1990; Rangdaeng et al, 1991). Although myoglobin is specific for skeletal muscle, its sensitivity for the diagnosis of rhabdomyosarcoma is low, particularly in poorly differentiated forms of the tumor (Tsokos, 1994). An alternative approach for the identification of cells with skeletal muscle differentiation involves the use of myogenic regulatory proteins (Li and Olson, 1992). Myogenic regulatory proteins play a key role in the commitment of primitive mesenchymal cells to a skeletal muscle lineage. Since these proteins are expressed earlier than structural proteins such as actin, myosin and desmin, they are particularly useful for the diagnosis of rhabdomyosarcoma. Antibodies to myoD1 and myogenin have been used extensively for this purpose (Wang et al, 1995). In exceptional cases, rhabdomyosarcomas may be positive for cytokeratins, neurofilament triplet proteins, neuron-specific enolase, S100 protein and leu 7.
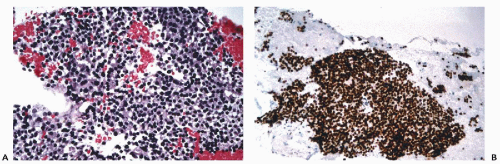 Figure 45-7 Metastatic poorly differentiated thyroid carcinoma in the lung. A. Cell block of fine needle aspiration biopsy stained with hematoxylin and eosin. B. Immunoperoxidase stain for thyroid transcription factor-1 in cell block demonstrates nuclear positivity. |
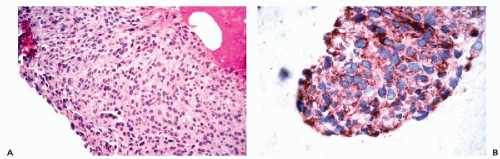 Figure 45-8 Metastatic leiomyosarcoma of the bladder involving the sacrum. A. Hematoxylin and eosin stain of cell block. B. Immunoperoxidase stain for smooth muscle actin in cell block demonstrates cytoplasmic positivity. |
tumors with endothelial differentiation, it is expressed in a variety of other tumor types (Ordonez, 1997). More recently, antibodies to CD31 and CD34 have been used for the identification of endothelial differentiation (Miettinen et al, 1994; von der Rijn and Rouse, 1994). It should be recognized, however, that a variety of soft tissue neoplasms, including dermatofibrosarcoma protuberans, solitary fibrous tumor, GI stromal tumors and peripheral nerve sheath tumors are also reactive for CD34. CD31, on the other hand, may be expressed weakly in some carcinomas and mesotheliomas (Suster, 2000).
assessment (see Chap. 31). B-cell lineage is most effectively accomplished by the application of CD19, CD20, CD22, and CD45RA antibodies (Fig. 45-9). In addition to these markers, the diagnosis of B-cell lymphomas and their differentiation from reactive processes requires the demonstration of light chain restriction by staining for kappa and lambda light chains. Monoclonality is defined as a kappa:lambda ratio of more than 6:1 or a lambda:kappa ratio of more than 4:1 (Sneige, 1990). In addition to CD20, antibodies that are of particular value in the categorization of mature B-cell lymphomas include CD43, CD5, CD10, CD23, and cyclin D1. Positive staining for CD43 and CD5 is characteristic of small lymphocytic lymphoma and mantle cell lymphoma. CD23 is consistently expressed in small lymphocytic lymphomas but is present in less than 20% of mantle cell lymphomas and marginal zone lymphomas. Among lymphomas, CD10 expression is limited to those of follicular type while cyclin D1 positivity is characteristic of mantle cell lymphomas.
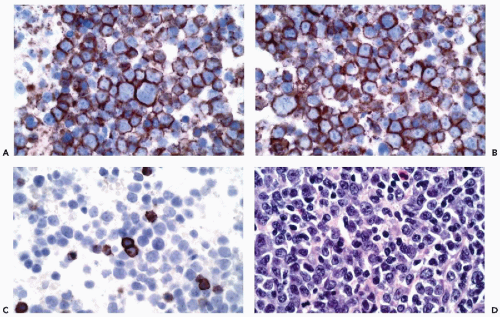 Figure 45-9 Large cell lymphoma. A. Immunoperoxidase stain for leukocytes common antigen (CD45) performed on a Papanicolaou stained aspirate demonstrates positive staining of the plasma membranes. B. Immunoperoxidase stain for CD20 demonstrates a similar pattern of staining. C. Immunoperoxidase stain for CD3 demonstrates a population of non-neoplastic T-cells. D. Hematoxylin and eosin stained section of a subsequent excisional biopsy demonstrates the typical features of a large cell lymphoma. |
tumors of nonepithelial type, such as paragangliomas and neuroblastomas, are positive for neurofilaments but negative for cytokeratins (Wick, 2000; DeLellis, 2001).
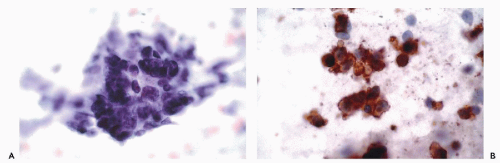 Figure 45-10 Small cell bronchogenic carcinoma. A. ThinPrep stained with Papanicolaou stain. B. Immunoperoxidase stain for neuron specific enolase on ThinPrep. The tumor cells were also positive for EMA and synaptophysin. |
While mature oligodendrocytes do not contain GFAP, cells that transiently express GFAP and myelin basic protein have been described. Staining intensity in glial tumors is inversely proportional to tumor grade (Schiffer et al, 1986; Morrison and Prayson, 2000). GFAP is also present in nonmyelinated Schwann cells, certain cells of the pituitary and breast and in tumors not considered to be of glial origin including mixed tumors of salivary gland and skin origin, nerve sheath tumors and chordomas.
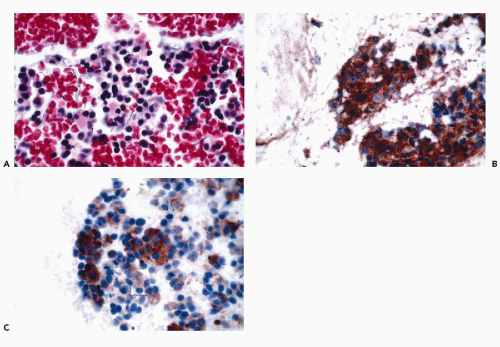 Figure 45-11 Pancreatic endocrine tumor. Cell block of fine needle aspirate stained (A) with hematoxylin and eosin, (B) with immunoperoxidase stain for chromogranin, and (C) with synaptophysin both showing cytoplasmic granularity. |
include angiomyolipoma and pulmonary lymphangioleiomyomatosis (Zamecnik, 1999).
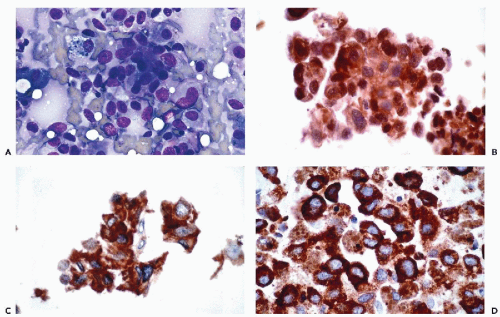 Figure 45-12 Metastatic malignant melanoma involving the lung. A. Diff-Quik stain. B. Immunoperoxidase stain for S100 protein demonstrates nuclear and cytoplasmic positivity. C. Immunoperoxidase stain for Melan-A (A103) demonstrates cytoplasmic positivity. D. Immunoperoxidase stain for HMB45 demonstrates cytoplasmic positivity. |



