Calcification Syndromes
When one considers the vast array of conditions in which human tissue can calcify, one can reasonably conclude that in addition to death and taxes, tissue calcification is the only real guarantee in life.
Calcification in tissue is seen in association with a wide range of disorders, including those in which there is a chronic serum elevation of calcium or phosphate; those in which connective tissue is abnormal; those seen following trauma, infection, and necrosis; and those as a component of tumors or arthritis (
1) (
Table 2.1). Most, if not all, clinical calcification syndromes can lead to ossification microscopically.
In general, ossification in tissue is seen when hydroxyapatite (HA), calcium pyrophosphate dihydrate (CPPD), and, to a lesser extent, other calcium-containing crystals (calcium acid phosphate, calcium carbonate, calcium oxalate, and calcium cholesterate) massively crystallize. This phenomenon requires, even in normal conditions of crystallization such as endochondral ossification, one or more of at least three phenomena: (a) local increase in the concentration of precipitable ionic species (e.g., Ca, PO4); (b) the presence of a “nucleator,” that is, a promoter of crystal deposition; or (c) the modification or removal of macromolecules inhibiting crystal deposition (
2).
Extracellular bone or soft tissue components that play a role in this process include collagen, proteoglycans, other noncollagenous matrix proteins, matrix vesicles, and their membrane-associated complexed acidic phospholipids (
3,
4).
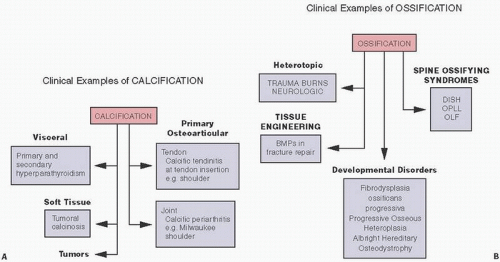
Clinical examples of calcification and ossification are protean (
Fig. 2.1).
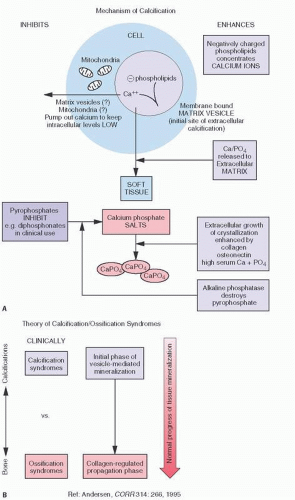
The mechanism of calcification is largely understood through observations of ultrastructural phenomenon. Membrane-bound bodies, called matrix vesicles, which contain lipid-rich membranes, are thought to be a key in this process (
5) (
Fig. 2.2). They pinch off from the plasma membrane of cells and migrate to the loose extracellular matrix space. Eventually, the lipid-rich membrane of these vesicles acts as a nidus for HA crystal formation.
Anderson (
5) has proposed a two-phase process: an initial phase of vesicle-mediated mineralization followed by a collagen-regulated propagation phase (
Fig. 2.2). Membrane-bound particles, called matrix vesicles, are thought to be key in this process. These 100-nm membrane-invested particles, which are located in both bone and cartilage, initiate calcification. Pinching off the outer plasma membranes of chondrocytes and osteoblasts, they generate HA crystallization. This initial phase is controlled by phosphatase enzymes such as alkaline phosphatase and other calcium-binding molecules. Phosphatases probably act by helping to produce a local increase in PO4, raising the (Ca+) × (PO
43-) product, resulting in initial deposition of mineral.
A second phase is one in which the matrix vesicles break down, exposing HA to the extravesicular milieu and a collagen-regulated propagation phase. Phase II is considered a physicochemical event regulated by nonenzymatic extracellular moieties such as collagen.
Matrix vesicle localization is specific. In the growth plate, it is along the longitudinal septae that preferentially calcify and, in bone, within newly formed osteoid (
5).
Rates of mineralization are affected by levels of Ca and PO4 in the extracellular fluid, pH, and controlling molecules such as proteoglycan and calcium-binding noncollagenous matrix proteins. Once begun, the process can continue producing a large cluster of tiny crystals, eventually forming a recognizable mineral mass. In the process, the matrix vesicles disappear. Termine (
6) has described this process as the “suicide mission” of matrix vesicles. The process of calcification is seemingly ubiquitous in human tissue and can be noted in vessels with aging, heart valves, following infections such as tuberculosis, and following trauma to soft tissue. What seems to control the seemingly inevitable process of calcification is the availability of protein inhibitors such as noncollagenous proteins and pyrophosphate. The complex of matrix vesicle-induced calcification is assisted by enzymes such as alkaline phosphatase and carbonic anhydrase. In addition, other lipid-containing proteins may be significant.
In order for crystalline material to form, being relatively insoluble, ions or groups of ions must collide with sufficient energy and proper orientation to form a critical nucleus, that is, the smallest stable structurally organized crystalline material that can persist in solution (
7). Nucleation and its subsequent growth can be promoted by increasing the activity of the ions, increasing diffusion rates, or removing inhibitors.
Numerous factors control calcification. For example, extracellular molecules (such as collagen and, in addition, noncollagenous proteins, proteoglycans, glycoproteins, and proteolipids) and smaller molecules and ions (such as pyrophosphate, carbonate, and certain hormones) regulate the production of a calcified matrix. Enzymes such as alkaline phosphatase and inorganic pyrophosphatases remove calcification inhibitors, allowing calcification to proceed. The entire process is thought to be controlled in part by certain cells such as osteoblasts and chondroblasts. Principal within cells controlling calcification is the mitochondria, which can accumulate calcium and inorganic phosphate and serve as a cytoplasmic reservoir or sink for calcium. For example, in the epiphysis prior to calcification, mitochondria within the chondrocyte become increasingly laden with calcium and phosphate.
In calcification involving cartilage, such as that seen at the epiphyseal growth plate, the process can be summarized as follows (
7): the chondrocytes secrete the collagenous matrix, which forms the framework for later mineral crystal deposition. They also produce proteoglycans, which consist of a protein core to which chains are attached and serve as an inhibitor of growth and nucleation of HA crystals, where such progression of mineral deposition is not desired. Modification of proteoglycan structure may permit calcification to proceed. The availability of calcium and phosphate to a certain extent is controlled by enzymes. Calcification inhibitors such as pyrophosphate ATP and other proteoglycan aggregates are also present and, in turn, are most likely under hormonal control. Thus, hormones control the concentration of ions in the extracellular matrix. Mitochondria act as a reservoir for calcium and phosphate to accumulate, and, extracellularly, matrix vesicles act as sites for initial mineralization deposition. Cells secrete the extracellular protein collagen, which provides the framework for subsequent apatite crystal deposition.
Variations of the phenomenon of calcification occur in growth plate mineralization such as normally occurring in the epiphysis, the dystrophic calcification that occurs in damaged tissue, the calcifications that occur presumably in association with some underlying matrix abnormality such as the collagen disorder scleroderma, and inflammation-related calcification as seen in dermatomyositis. Following trauma, one may see poorly organized, amorphous looking extracellular soft tissue deposition of HA crystals or calcium phosphate crystals in what are termed dystrophic calcification syndromes. In other instances, trauma is followed by the actual deposition of bone, that is, heterotopic ossification (HO) or myositis ossificans (MO) circumscripta. What key events dictate the direction in which mineralization will go, whether on to either calcification or bone formation, remains an enigma.
There is an overlap in the microscopic observation of calcification and ossification in these disorders. For example, in tumoral calcinosis, which is characterized by hyperphosphatemia, the characteristic deposits are calcium phosphate in the soft tissue, but in histologic section, there may be actual bone formation (
Fig. 2.3). Normally, calcium and inorganic phosphate are present in serum or extracellular fluid at concentrations that are too low for spontaneous precipitation but of a sufficient quantity to cause HA or amorphous calcium phosphate deposition once the crystal nucleation process has begun. The normal presence of inhibitors such as inorganic pyrophosphate is necessary to prevent this in the normal situation.
Bone formation, however, requires the presence of extracellular collagen matrix. In normal bone, this requires a finely organized structure. Bone collagen is highly insoluble because of many covalent
intermolecular and intramolecular cross-links and is predominantly that of type I collagen. In bone matrix, collagen is packed with short gaps in a one-quarter stagger array. HA is thought to deposit within these gaps. In addition to type I collagen, which serves as a scaffold for bone formation, bone mineral nucleators must be present. These phosphoproteins include a wide array of most anionic macromolecules that can bind Ca
+ in solution or on the apatite crystal surface (
Fig. 2.2).
Enzymes that have been associated with the mineralization process include those that regulate phosphoprotein phosphorylation and dephosphorylation. Of these, alkaline phosphatase has been extensively studied. It hydrolyzes phosphate esters, thereby increasing phosphate concentration, and this enhances mineralization. Alkaline phosphatase may also remove phosphate inhibitors of apatite growth such as ATP. The clinical syndrome of hypophosphatasia, which is characterized by a deficiency of alkaline phosphatase, shows marked abnormal bone mineralization. These puzzling biochemical factors do help in understanding the morphologic differences between woven and lamellar bone. Woven bone (fibrous bone, immature bone, embryonic bone) has more cells and collagen than that of the mature adult skeleton. In addition, the collagen has a coarser appearance on light microscopy, and there is an irregular orientation with random osteocyte distribution, creating a haphazard appearance on polarized light microscopy (
Fig. 1.7). In the normal adult skeleton, there is little woven bone, although it may be seen near tendon and ligament attachments and near cranial sutures. Woven bone is the hallmark of Paget’s disease (the mosaic bone), but is a nonspecific finding and may be seen in fracture repair, with certain bone tumors, and in hypermetabolic states such as hyperparathyroidism. Although the appearance of matrix vesicles in woven bone is more obvious than in lamellar bone, the presence of woven bone in disorganized hypermetabolic conditions suggests that the basic multicellular unit and its dynamics may be interrupted in this process.
Lamellar or mature bone consists of discrete alternating bands approximately 2 to 3 µm in thickness, revealing a high order to the collagen bundles. There is a more even distribution of osteocytes, and they are flat as opposed to spheroid in woven bone. Lamellar bone is formed more slowly than woven bone.
Clinical Syndromes
Hypercalcemia or hyperphosphatemia of any etiology (
Table 2.1) can lead to supersaturation and, thus, HA crystal deposition.
So-called metastatic calcifications are predilected to the kidneys, lungs, the media of large arteries, conjunctiva, periarticular soft tissue, endocardium, and fundus of the stomach.
In dystrophic calcification syndromes, calcinosis usually occurs in the skin or just below it. It may be relatively localized, especially over extensor aspects of joints and fingertips (calcinosis circumscripta), or more widespread in subcutaneous tissue (calcinosis universalis).
The prototype metastatic calcification syndrome due to hypercalcemia is primary hyperparathyroidism (see
Chapter 4).
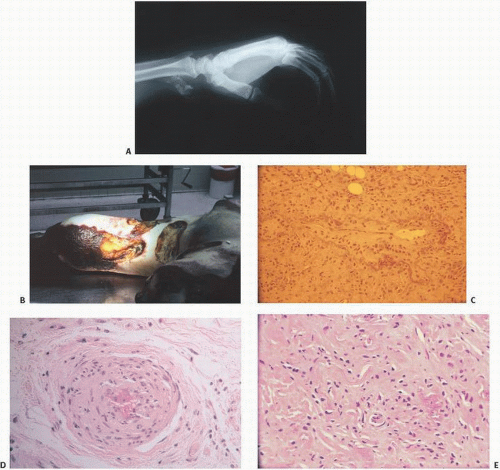
The histopathology of the calcific deposits in virtually all calcifying syndromes is identical to a variable pattern of aggregates of amorphous poorly refractile crystalline deposits that have a granular appearance (
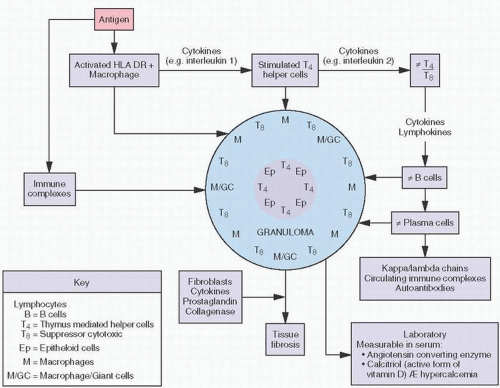
Fig. 2.3A). Calcific deposits may be associated with peripheral inflammation or even multinucleated giant cells, giving rise to the term granulomatous calcification, a condition characterized by the local recruitment of macrophages to attempt to remove the deposits (
8) (
Fig. 2.4).
Sarcoidosis
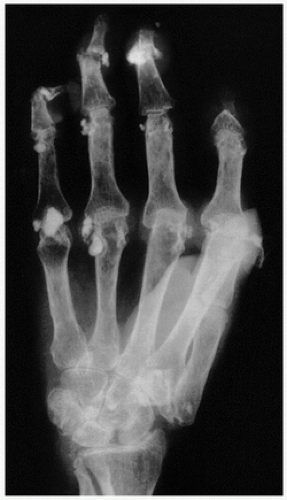
Sarcoidosis is a systemic disease of unknown etiology characterized pathologically by the presence of noncaseating granulomas. In about 15 percent of cases, there may be transient hypercalcemia probably resulting from increased plasma concentrations of 1,25-vitamin D (
Fig. 2.5) (
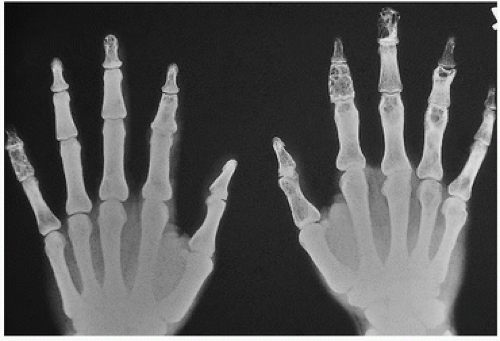
9). This hormone increases dietary absorption of calcium and enhances bone resorption. Hypercalcemia may eventuate into roentgenographically detectable calcifications. Characteristically, the small bones in the hands and feet are involved, giving a fine, reticular pattern (
Fig. 2.6). Occasionally, cysts may be seen. Total destruction of the phalanges may be seen in advanced states (
10).
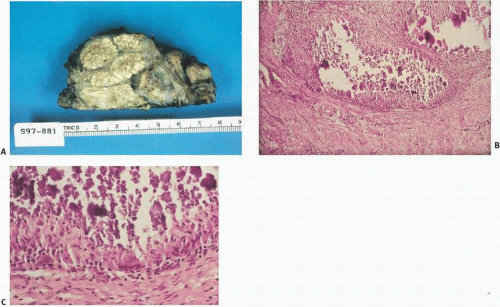
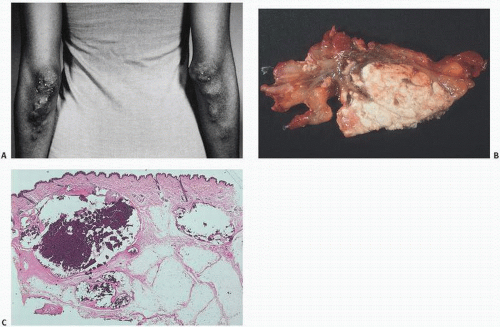
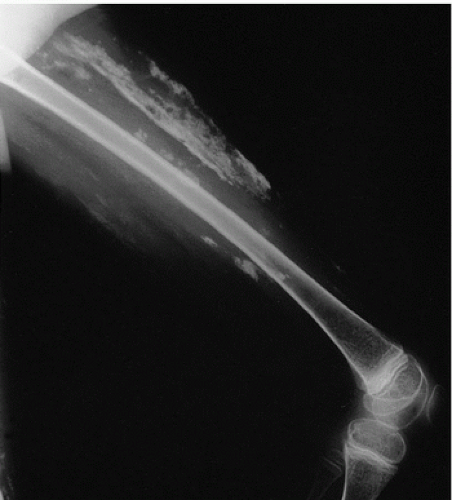
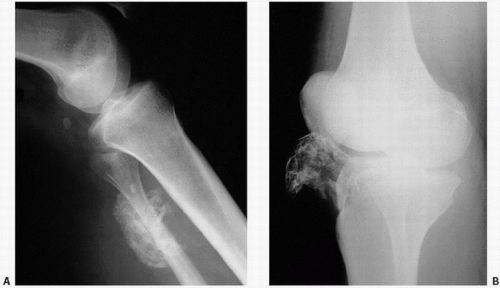
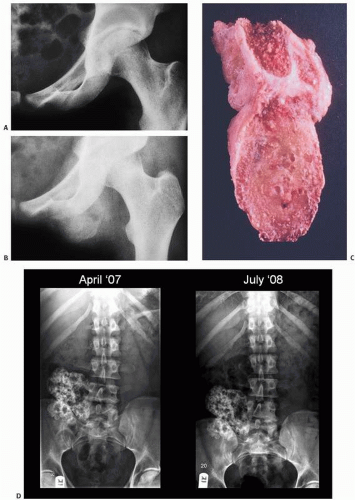
Tumoral Calcinosis
Tumoral calcinosis, first described by Inclan (
11) in 1943, is a rare disease characterized by the formation of large, painless, juxta-articular calcific masses, often at pressure points (
Fig. 2.7). Both nonfamilial and familial types of tumoral calcinosis have been described. The familial type (OMIM#211900, online Mendelian inheritance in man) is further divided into hyperphosphatemic and normophosphatemic types. Several mutations have been found in the hyperphosphatemic familial type (HFTC) involving inactivating mutations in UDP-N-acetyl-α-D-galactosamine including:
3(GALNT3),
FGF-23,
KLOTHKO.
Mutations in the normal phosphatemic form include those involving sterile α-motif domain-containing protein 9 (SAMD-9).
The calcifications in tumoral calcinosis are typically slow-growing and painless. Clinical complications accrue when the masses infiltrate the skin or other tissues. Pressure point sites such as the elbows and buttocks may be preferentially involved in a bilateral distribution.
Radiographs reveal large diffusely radiodense lobulated masses (
Fig. 2.8).
Tumor calcinosis is an HA deposition disease.
Grossly, the lesions of tumor calcinosis have a gritty texture but large lesions may consist of a calcareous material in a viscous and milky fluid. Histopathologically, the aggregates of HA mineral are nonrefractile and may be associated with histiocytic macrophages and multinucleated osteoclast-like giant cells. Patchy osseous transformation may occur.
Electron microscopic investigations have shown that nonbound intracellular crystal deposition exists, especially in the vicinity of dilated granular endoplasmic reticulum of cells that contain abundant amounts of that organelle. The marked accumulation of intracytoplasmic fine filaments, independent of crystal deposits, might indicate a state of heightened metabolic activity associated with an active or degenerative change.
The x-ray microanalysis findings have shown the presence of calcium and phosphorus with an average atomic ratio of 1.54 for Ca/P.
Electron diffraction patterns obtained from many discrete crystal deposits as well as x-ray diffraction analyses have all indicated that the crystals in these deposits are larger and/or more perfect than those observed in bone. Deposits are composed of apatite mineral. The collagen content of the tumoral calcinosis deposits is lower than that of bone.
The uronic acid and total lipid contents of the matrix found in the deposits are closer in value to those of the surrounding tissues than to those of bone.
Treatment of tumoral calcinosis can be difficult and frustrating (
12). Surgical excision has been associated with high recurrence rates but may relieve pain, improve function and cosmesis.
In general, in the hyperphosphatemic forms, discontinuation of supplemental phosphate and hydration will help renal clearance. Dietary phosphate restriction and diuresis may help. Aluminum hydroxide in combination with dietary phosphate and calcium restriction has been used as the phosphate-binding agent sevelamer.
The understanding that tumoral calcinosis deposits are better mineralized than bone and are quite different from bone clarifies
some of the difficulties associated with treatment of this disease. Treatment by phosphate binders to reduce the plasma phosphorus levels has not been universally effective. In vitro, the large apatite crystals of tumoral calcinosis deposits, like the smaller crystals of bone mineral, have been shown to grow and proliferate in synthetic lymph solutions that have normal serum phosphate levels as well as in solutions in which serum phosphate levels are reduced by 50 percent. Thus, lowered serum phosphate levels would not be expected to remove the deposits, although lowering the serum phosphate level should reduce the incidence of formation of new masses and the rate of growth of existing masses.
Renal Disease
In renal disease, soft tissue calcification can be profound and is probably due to many factors (
Chapter 4). Ultrastructural changes similar to tumoral calcinosis occur: HA crystal formation and calcification developing primarily from intracytoplasmic membrane-bound vesicles and also from mitochondria. Factors include elevated phosphorus and/or calcium above physiologic saturation points, secondary hyperparathyroidism, problems in the metabolism of vitamins D and K and magnesium, alkalosis, and focal tissue injury. Aluminum intoxication—a known interloper in dialysis treatment and a substance that blocks mineralization—may also precipitate a calcium efflux from bone. Oxalosis (see
Chapter 4) is also associated with renal disease.
Cutaneous Calciphylaxis
Cutaneous calciphylaxis, also called calcific uremic arteriolopathy, is a serious complication occurring in as many as 4 percent of patients with terminal renal failure. Seen more in women than men, the clinical features include hyperparathyroidism (80 percent), hypercalcemia (20 percent), and hyperphosphatemia (68 percent) (
13). Characterized by painful and pruritic skin lesions, violaceous and indurated nodules progress to skin necrosis, ulceration, and eschar formation.
Suspect the disease in renal patients when there is hypercalcemia, hyperphosphatemia, elevations in the calcium-phosphate product, hyperparathyroidism, and exposure to calcium and vitamin D products that when used to suppress parathyroid hormone levels can lead to higher levels of both calcium and calcium phosphate products.
Often subtle even microscopically, endovascular fibroblastic proliferation, wiry vascular luminal calcification, calcifying septal panniculitis, and, in late stages, epidermal ulceration and dermal necrosis are seen (
Fig. 2.9) (
14) (see
Chapter 4). It has been associated with mortality rates as high as 60 to 80 percent.
Disorders in which soft tissue calcification occur in the setting of normal serum calcium and phosphorus are summarized in
Table 2.1.
Dermatomyositis
Dermatomyositis is a multisystem disorder characterized by vasculitis involving small vessels usually of the skin and striated muscle. It is more common in women and is seen both in childhood and adulthood. Soft tissue calcifications occur several years after the onset of the disease and may or may not progress. Spontaneous resolution has occurred. Depositions occur usually in periarticular regions or areas subjected to trauma. Calcifications may be more localized (circumscripta) usually around joints. Symptoms may include pain, limitation of mobility, and skin ulceration. The finding of increased urinary γ-carboxylated proteins has suggested that calcium-binding protein may be the precipitating event. Because calcinosis occurs infrequently in long-term survivors, some believe it to be a scarring phenomenon.
The radiographic presentation of dermatomyositis is characterized by soft tissue calcifications, usually located between muscle planes or at the junction of subcutaneous fat with muscle groups or skin (
Fig. 2.10). These calcifications tend to follow anatomical planes. Treatment at times can result in resorption of these soft tissue calcifications.
Osteoporosis is found in most cases of dermatomyositis. The osteoporosis tends to be periarticular. Small erosions are unusual findings in the joints involved by dermatomyositis, as well as in the phalangeal tufts. Radial dislocation or subluxation of the interphalangeal joint results in a “floppy thumb,” a finding not unusual in dermatomyositis. Subcutaneous edema and muscle edema can cause increased bulk, which is sometimes detected by radiographs.
The prognosis in dermatomyositis in children is variable, but may result in joint contractures and weakness.
Scleroderma (Systemic Sclerosis)
Scleroderma (systemic sclerosis) is a complex disease characterized by fibrosis, vascular alterations, and the presence of autoantibodies such as antinuclear antibodies and anti-polymyositis-scleroderma. The soft tissue calcifications seen in association with scleroderma are a relatively common finding occurring in up to one-quarter of patients (
15). They usually appear late in the disorder and are more common in women, particularly in the skin. The calcifications are typically focal and most frequently seen in the hands (
Fig. 2.11).
Other locations include over the knees and elbows, paraspinous and vertebral disc calcification, intraspinous calcification, and the foot. In the CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasis), calcification is one of the defining elements of the clinical picture (
15).
Posttraumatic Calcification
Perhaps the most common type of dystrophic calcification is that following trauma. Although inflammation of soft tissue may be associated with radiographically detectable calcium deposits, the etiology of calcification in this setting, although commonly observed clinically, is not clearly understood.
Most likely, the calcification is due to injury-induced small-vessel thrombosis and ischemia followed by myonecrosis (vida infra). The calcium salts are nonrefractile HA crystals, which, similar to other HA disorders, can lead to granulomatous-like multinucleated giant cell reaction. Specific eponymous conditions have been described such as the Pellegrini-Stieda syndrome, where soft tissue calcifications follow injury to the medial collateral ligamentous complex of the knee.
Milwaukee Shoulder
The association of a large rotator cuff tear and damage to the glenohumeral joint, which may be accompanied by basic calcium phosphate crystals and active collagenase in the joint fluid, has been termed the Milwaukee syndrome (
16). It has been postulated that the crystals are the culprits and the tissue damaged as a result of a crystal-induced arthropathy. Mostly, these deposits are characterized by a milky emulsion, which, after dehydration, becomes a fine granular powder. Histopathologically, these lesions are characterized by amorphous, well-circumscribed or poorly circumscribed deposits of mineral material, nondescriptive but appearing as dark staining on routine hematoxylin and eosin preparation. There may or may not be associated inflammation.
By x-ray diffraction analysis, these calcific deposits contain HACA10 PL46 OH2 as the only inorganic constituent. The diffraction pattern demonstrated poorly crystallized HA, that is, small crystalline particles similar to those of bone. The sometimes observed presence of active phagocytosis may be linked to disintegration of the bonding capacity of the organic molecule (
17).
Calcific Myonecrosis
Calcific myonecrosis is a phenomenon that typically occurs in the lower extremity following either a compartment syndrome or vascular or neurologic injury (
18). The injury may predate clinical detection by many years, often 30 years or more.
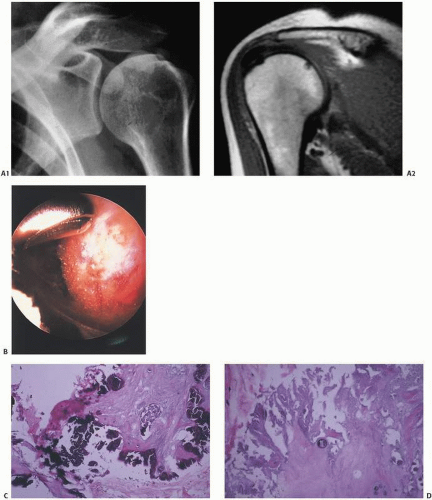
Calcific Tendinitis
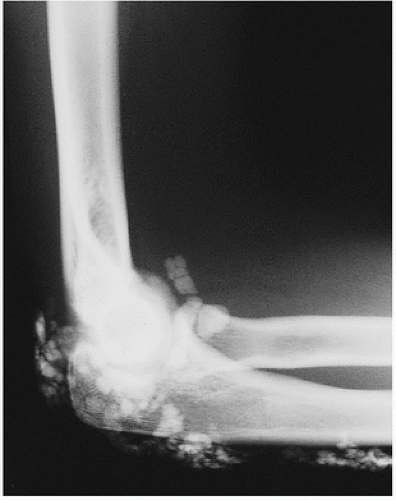
Although virtually any capsular, tendinous, ligamentous, and bursal tissue may be the site of calcific deposits, those of the shoulder are the most commonly observed clinically (
Fig. 2.12). The supraspinatus tendon has received the most attention, and theories of its origin range from inflammation and necrosis to focal metaplasia, the latter proposed to be due to poor oxygenation of the poorly perfused distal region triggering chondroid metaplasia and subsequent calcification.
Calcifications usually occur within a tendinous insertion, but they may affect peritendinous tissue and may be difficult to precisely locate their origin. T
2-weighted magnetic resonance imaging (MRI) images are usually high signal due to the related inflammation. There may be erosion of cortical bone, but extension through the bone into marrow would be rare (
19).
Clinically, calcific deposits in calcific tendinitis can spontaneously resolve within 6 to 10 weeks.
Calcified Tumors
Roentgenographically detectable calcification and even ossifications have been seen in a wide range of tumors, both skeletal and nonskeletal in origin (
Fig. 2.13). Calcifications have been described at least microscopically in virtually every soft tissue tumor. Notable exceptions in the orthopaedic literature are the giant cell tumor of tendon sheath and pigmented villonodular synovitis (PVNS). Ossification in tumors has also been noted in a wide range of disorders (
20). Calcifications are most commonly observed in tumors, benign and malignant, of cartilaginous origin (
21).
Calcium Pyrophosphate Crystal Deposition
In addition to the aforementioned causes of extraskeletal calcification, the differential diagnosis should also include rare circumstances in which CPPD deposition occurs in a massive tumorlike form that is most often limited to a single joint. Patients usually present with a firm, juxta-articular mass, which may or may not
be painful, and may produce limitation of motion or neuroplexus syndrome. There is often progressive increase in size with or without attacks of pseudogout. Roentgenographically, juxta-articular, lobulated, radiodensities are noted. The diagnosis is established by identifying the classic positively birefringent crystals on crystal analysis of fluid aspirates from the joint. Patients usually have no other joint involvement by CPPD.
Intervertebral Disc Calcification
Intervertebral disc calcification (IDC) is a common finding on radiographic imaging, the prevalence of which has been reported in up to 5 percent on chest radiographs and 6 percent on abdominal radiographs (
22). It increases with age and the extent of disc space loss and, in the elderly is more frequently seen in the annulus fibrosus and in the lower thoracic spine.
IDC in the pediatric population is more often painful and more often seen in the nucleus pulposus of the cervical spine. There may be torticollis, fever, leukocytosis, and elevation of the sedimentation rate. The etiology is unclear. Prognosis is excellent with resolution of pain with conservative treatment in most cases.
Musculoskeletal Calcifications
Musculoskeletal calcifications can be seen in a miscellaneous group of other conditions. Anything leading to fat necrosis can evolve into calcification, which may explain entities such as prepatella bursitis. Vessels calcify with age.
Many drugs, such as the chelator calcium disodium edetate, can lead to tissue calcification. Recently, drugs such as gefitinib and denosamaub have caused osteosclerotic changes. Gefitinib, an oral drug that inhibits receptor tyrosine kinase, including the epidermal growth factor receptor, in the treatment of lung cancer (
23), and denosamaub, used in the treatment of osteoporosis, giant cell tumor, and PVNS, have had osteosclerotic effects. Such effects may mark a favorable outcome in treatment.
Heterotopic Ossification
HO is defined as bone formation in nonosseous tissue. The formation of bone in soft tissue has been established in many experimental and clinical settings, and the induction of bone is thought to result from the ability of pluripotent mesenchymal cells to differentiate into osteoblasts, which in turn initiate HO. In chicken embryonic membranes, a well-developed model, bone formation depends on the interaction between the mesenchymal tissue and epithelium or a component of the basement membrane. Induction may also be mediated by nonliving cellular tissue as in bone grafts or recently investigated material such as bone-demineralized protein (
24). It is now generally thought that the conversion of progenitor cells to osteogenic precursor cells as the result of cell-mediated interactions with the local tissue environment and contributions by oxygen tension, pH, availability of micronutrients and mechanical stimuli leads to heterotopic ossification (
25). The most investigated “inductive” substance has been bone morphogenetic protein (BMP), a polypeptide or group of polypeptides thought responsible for postfetal differentiation of mesenchymal cells into cells (chondrocytes and osteoblasts) capable of forming bone at ectopic sites by endochondral ossification. The association of heterotopic bone formation with various epithelial tissues such as urinary tract epithelium has suggested that the epithelium adjacent to developing mesenchymal cells may influence the differentiation of mesenchymal tissue toward ossification. Indeed receptors in the matrix extracellularly may modify these effects. Mesodermal cells, in fact, may have a predetermined, inherent ability to differentiate along bone and cartilage lines when inductor factors unleash this potential.
There are numerous tissues in experimental settings that have led to the induction of bone formation, and these include human fibroblasts, certain human epithelial tumors, and human amnion. Clinical situations also abound in which the formation of bone is readily demonstrable (
Table 2.2). These include HO or “MO” following trauma; heterotopic bone formation following certain
orthopaedic procedures such as joint replacement, particularly of the hip; bone formation following bone marrow transplantation; and of particular interest from an investigational point of view, the formation of bone following implantation of demineralized bone matrix (DBM) initially described in 1965 by Urist. In his initial experiments, bone demineralized by hydrochloric acid implanted within muscles in allogeneic laboratory animals stimulated mesenchymal cells to proliferate and differentiate into chondroblasts and osteoblasts, with the initially formed cartilage developing into bone. Several weeks into the experiment, bone and bone marrow appeared.
Not surprisingly, HO has been well described as a complication in the use of recombinant BMPs, commercially available osteoinductive material such as Infuse and OP-1, available for the management of fractures and spinal fusion procedures (
25).
Clinically observed processes of ossification include that by epithelial tissue such as gastric mucosa and seminal vessel mucosa; osteoinduction by chronic inflammation; heterotopic mesenteric ossification (
26); and ossification occurring in postoperative abdominal scars, cancer, and even phleboliths.
In addition to general trauma, three important clinical precursors to HO are burns, neurogenic injury, and major surgery such as a total joint replacement.
HO is a well-known complication of burns and may be observed in as many as 30 percent of patients. The elbow is often the involved site in thermal burns, and a delay of 18 to 24 months before surgical excision has been recommended to avoid reoccurrence (
27).
Neurogenic HO is the occurrence of ectopic bone in soft tissue and periarticular locations following marked injury to the brain or spinal cord (
28). It has also been well documented in nontraumatic neurological diseases such as Guillain-Barre syndrome, tumors and infections, and hemorrhages of the central nervous system.
Neurogenic HO is more common in men and younger patients.
Although its etiology is unknown, proposed pathogenetic mechanisms have included the vasomotor, vascular stasis, and metabolic and trophic disorders that follow prolonged immobility and the possibility of the induction of endochondral ossification by the forcible passive mobilization of the paralyzed limb during rehabilitation, which may lead to repetitive tendon and muscle trauma (
28), which may lead to the proliferation of cells with osteogenic potential.
The formation of bone following orthopaedic procedures such as total hip replacement is a frequently observed disorder that is characterized by initial inflammatory lesions within muscles and subsequent deposition of bone. Ahrengart and Lindgren (
29) defined the patient at risk (
Table 2.3). It is thought to complicate total hip arthroplasty in up to 67 percent of cases with severe disability (5 to 31 percent). HO has been reported after virtually every joint
replacement including total knee replacement (4 to 40 percent), revision knee replacement (56 percent), intervertebral disc replacement (4.3 percent), shoulder and elbow replacement, and ankle replacement (35 percent).
The etiology may be the transformation of inducible osteoprogenitor cells in response to bone-stimulating proteins released from damaged muscular tissue. Because it has also been described without a history of trauma, there may be a genetic predisposition. Heterotopic bone formation following total hip replacement is most common following arthroplasty for osteoarthritis and may be related to the type of osteoarthritis. Those with hypertrophic degenerative joint disease may be more susceptible, an intriguing possible link between osteophyte productions in degenerative joint disease and heterotopic bone.
The following risk factors for HO following total hip arthroplasty have been identified: previous HO, hypertrophic osteoarthrosis, ankylosing spondylitis, inferomedial osteoarthrosis, posttraumatic osteoarthrosis, direct lateral operative surgical approach, Paget’s disease, and diffuse idiopathic skeletal hyperostosis (DISH).
The biochemical basis for the development of heterotopic calcification is not clear, but numerous data point to the concept that interaction between grafted cells and the host mesenchymal tissue is necessary for the induction of bone to take place. BMP factors may be important. Biochemical markers for osteoinductive activities, such as alkaline phosphatase, may be high initially, but can be normal in the maturing lesion. An elevated alkaline phosphatase level in an atypical clinical or radiographic setting should raise the suspicion of an extraskeletal osteosarcoma.
Factors that may inhibit, either clinically or experimentally, the development of osteogenesis include aging (in which osteogenesis declines) and iatrogenic injuries to collagen synthesis such as that induced by penicillamine and corticosteroids, probably through suppression of potential cell proliferation. Vitamin D3 analogs such as dihydroxycalciterol and calcitonin may induce osteogenesis. Aluminum also adversely affects endochondral bone formation.
Finally, heterotopic bone is similar in biochemical and biologic properties to normal bone with the exception of the lack of a periosteal membrane and, thus, most likely a limited capacity for regeneration.
Focal transformation of bone can be observed histologically in virtually every connective tissue calcifying syndrome (calcinosis syndromes, tumoral calcinosis), in a host of benign tumors, and even as an accompanying phenomenon to illnesses as diverse as infection and atherosclerosis, in which ossification of the coronary arteries and aorta is well established. One may ponder whether a natural end stage of human tissue differentiation is bone.
Traumatic Heterotopic Ossification
Myositis Ossificans (Circumscripta or Traumatica)
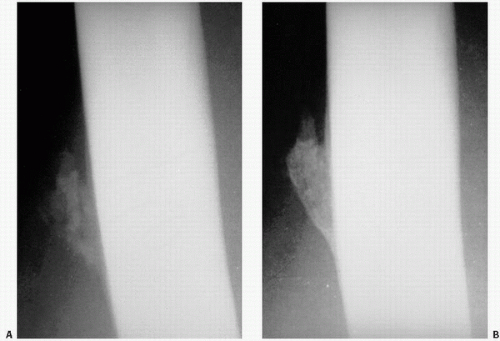
It has long been observed that following blunt trauma to the body, swelling in soft tissue may be followed roentgenographically by the development initially of a vague shadowlike density and, eventually, a well-defined peripherally ossified mass (
Fig. 2.14). This clinical picture has come to be known as myositis ossificans or myositis ossificans traumatica (
30). The term, coined by Von Dusch in 1868, is misleading in that muscle is not necessarily involved. The term heterotopic ossification is more appropriate.
A plethora of experimental evidence consistently observed preceding clinical events suggests that HO is the end result of a wide array of inciting injuries (
31) including college fraternity hazing (
32) and a wide range of bodily blunt trauma in competitive contact sports. The chronology of HO has been described (
Table 2.4).
A large number of osteoblast or osteoblast-like cells, present in the periosteum, cortical bone, endosteum, trabecular bone, and marrow stroma, once activated, can form new bone. Inducible osteoprogenitor cells include fibroblasts, endothelial cells, perivascular cells, lymphocytes, satellite cells of striated muscle, bone marrow stromal cells, and undifferentiated mesenchymal cells.
Classically, MO circumscripta once “matured” is a nonprogressive benign osseous bony lesion.
The patient usually presents with a lump in a muscle that has been present for some weeks and that may have been somewhat painful. It most commonly occurs in the thighs and arms and in soft tissue along the diaphysis of bones where repetitive trauma can be expected. Patients are usually 20 to 40 years old. Typically, there is painful swelling with rapid growth followed by shrinkage. The process lasts about 3 weeks, but edema can last several months. A roentgenogram taken soon after the onset of symptoms may not show an opacity, but within 1 or 2 weeks, a poorly defined shadow will appear, and over the following weeks, the periphery of this shadow will become increasingly well delineated from the surrounding soft tissues, the latter often an important distinguishing feature from true sarcoma. Typical MRI pattern may be hard to define, reflecting the protean manifestation of tissue change in MO. Classically, however, a low signal intensity-rim is a characteristic finding.
Clinical complications of HO leading to various states of disability include pain, joint contracture, ankylosis, spasticity, fever and swelling, neurovascular compression, lymphedema, and pressure ulcers (
33).
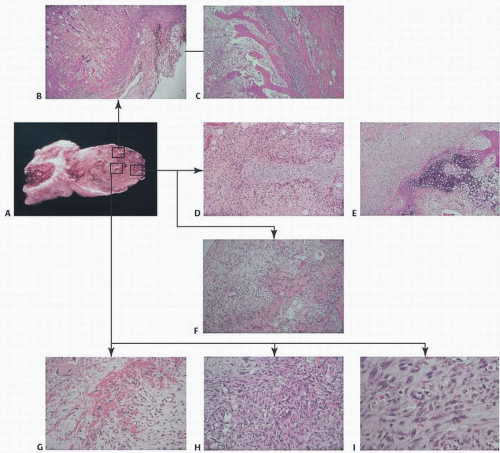
The gross examination of a focus of MO circumscripta present for 1 or 2 months will show a shell of bony tissue with a more or less soft reddish brown central area. It is usually 2 to 5 cm in diameter and adherent to the surrounding muscle (
Fig. 2.14C). The mass parallels the bone shaft along the axis of the muscle.
Microscopic examination reveals, in the central part of the lesion, an irregular mass of active immature fibroblastic cells with foci of interstitial microhemorrhage (
Fig. 2.15). Warfarin (Coumadin), which prevents clotting by inhibiting the hepatic synthesis of vitamin K-dependent clotting factors, may result in hemorrhage, which has been implicated as an inciting ossification trigger in this setting. Occasionally, hemosiderin-filled macrophages and degenerative muscle fibers are encountered. The whole lesion is intensely vascular, the vessels being dilated channels lined by endothelium, but without any formed media or adventitia. At some distance from the center of the lesion, depending on the age of the lesion in question, one finds small foci of osteoid production and, rarely, cartilage production. This tissue may be disorganized and hypercellular. As one approaches the periphery, there are more and more clearly defined trabeculae (
Fig. 2.15). The bone is usually of the primitive woven type with large, round, and crowded osteocytes; however, in cases of long standing, the bone is mature and has a lamellar pattern.
Histologically, it may be difficult to differentiate a focus of myositis, especially in its acute and active stage, from a sarcoma. Terms such as “pseudomalignant MO” underscore diagnostic dilemmas (
34). A careful correlation of the clinical and x-ray findings is therefore essential. An important distinction to be emphasized is that whereas MO is most mature at its periphery and least mature at its center, the opposite is true of osteosarcoma.
MO circumscripta may spontaneously regress. Some investigators equate the lesion with an ossifying hematoma. In contrast to osteogenic sarcoma, MO grows more rapidly and is more painful.
The treatment of choice in MO is observation because most lesions become and remain asymptomatic and eventually regress. Operations should be avoided because surgical intervention may reactivate the disease. Nevertheless, diagnostic uncertainty, limitation of range of motion, and impingement on neurovascular tissue may require intervention. Some advocate delaying surgery for 6 months after the initial trauma to allow the bone to mature and form a fibrotic capsule. Interestingly, fractures through MO tissue, if treated without surgical intervention, can heal without re-exacerbation of the disease (
35).
Various approaches that have been used to prevent HO, especially for total hip arthroplasty, include gentle passive range of motion techniques and physiotherapy; preoperative and postoperative radiation; and anti-inflammatory medications such as indomethacin, bisphosphonates, colchicine, calcium antagonists, and Coumadin (warfarin-type) anticoagulants. Radiation therapy and nonsteroidal anti-inflammatory drugs (NSAIDs), two distinct treatment options, are currently the main considerations, with little evidence supporting one over the other in preventing HO.
Radiation prophylaxis has been used both preoperatively and postoperatively with 800 cGy (
36). Experimental results in a rabbit model have supported this approach (
37). Preoperative radiation is an effective approach. Single-dose radiation therapy has been well tolerated.
Currently, NSAIDs are the leading choice for prophylaxis. Usually given perioperatively, there is less evidence to support postoperative effectiveness. Nonsteroidal anti-inflammatory agents including cyclooxygenase 2 (COX-2) inhibitors have been found effective after total hip replacement. A risk of long-bone nonunion after 6 weeks of systemic indomethacin has been observed (
38).
Imatinib mesylate (Glivec), a drug known to inhibit platelet-derived growth factor (PDGF), which plays a role in angiogenesis (an initial phase of HO), has been used with some success, emphasizing the ongoing efforts to creatively block the HO cascade (
33).
Novel experimental approaches to inhibit HO in animal models have included the retroviral delivery of Noggin, a BMP antagonist. This suggests that manipulation of the microenvironment in tissue may be the key to successful treatment (
39).
Avulsion of apophyses and epiphyses, especially in the young individual, can lead to similar calcification and ossification in the soft tissues. The initial presentation can be confused, from the radiologic point of view, with a bone-producing malignancy, an osteosarcoma, especially the parosteal or juxtacortical type. Most areas of MO progress as a soft tissue mass, which is denser in the periphery and lucent in the center, different from osteosarcomas, which tend to be denser in the core or center of the lesion. The awareness of preferential locations for avulsion of ossification centers is also helpful in the differential diagnosis. These anatomical sites prone to avulsion of the ossification centers include the lesser trochanter (avulsion by the iliopsoas), greater trochanter (avulsion by the abductor muscles), ischial apophyses (avulsion by the hamstrings), lateral surface of the iliac wing above the edge of the acetabulum (avulsion by the reflected head of the rectus femoris), iliac crest (avulsion by the quadratus lumborus), and the symphysis pubis (avulsion by the adductors). The rapid change in radiographic appearance is also a finding common in the initial stages of MO (
Fig. 2.16).
HO has been well documented following high-energy blast injuries sustained in modern warfare and has been estimated to occur in approximately 64 percent of those patients (
40). Since modern blast injuries are usually characterized by massive zones of injury and prolonged systemic inflammation, the sheer size of HO involvement in these soldiers should not be surprising.
Although some patients remain asymptomatic, others experience transiently painful symptoms. Mimicking classic localized blunt trauma HO, early inflammatory phases transition to a more quiescent maturation phase.
Clinical studies of combat wound HO have demonstrated that injured patients have significantly higher numbers of muscle-derived connective tissue progenitor cells, and those developing HO, higher numbers of assayable osteogenic colonies. Both of these findings are considered important potential catalysts for the HO cascade (
40).
The presence of an amputation in these patients has been found to be independently associated with the development of HO (
41).