Shigella and enteroinvasive Escherichia coli
Paradigms for pathogen evolution and host–parasite interactions
Anthony T. Maurelli, Uniformed Services University, Bethesda, MD, USA
Background
Bacillary dysentery or shigellosis is caused by members of the Shigella genus and a group of pathogenic strains of Escherichia coli known as enteroinvasive E. coli (EIEC). Dysentery was the term used by Hippocrates to describe an illness characterized by frequent passage of stools containing blood and mucus accompanied by painful abdominal cramps. This disease has had a tremendous impact on human society over the centuries with perhaps one of the greatest effects being its powerful influence in military operations. Long, protracted military campaigns and sieges almost always spawned epidemics of dysentery and caused large numbers of military and civilian casualties. The Napoleonic campaigns and the American Civil War were 19th century examples of the devastation caused by dysentery. This capacity of dysentery to factor in the outcome of nation-shaping events continues in the modern era. Soldiers were stricken with the disease in essentially all major conflicts of the 20th century, including both World Wars, Korea, Vietnam, and the first Gulf War. Indeed, this ancient disease goes hand in hand with political and socio-economic strife as dysentery continues to claim the lives of hundreds of thousands of people living in the unsanitary conditions brought on by war and poverty. The low infectious dose required to cause disease coupled with oral transmission of the bacteria via fecally contaminated food and water, accounts for the spread of dysentery caused by Shigella spp. in the wake of many natural (earthquakes, floods, famine) as well as man-made (war) disasters. For example, refugees of a recent conflict in Zaire suffered high morbidity and mortality rates brought on by bacillary dysentery (Kim et al., 2009). Even apart from these special circumstances, shigellosis remains an important disease in developed countries as well as in underdeveloped countries. The global burden of shigellosis has been estimated at over 163 million cases per year with nearly 1 million deaths, the majority occurring in children less than 5 years of age (Kotloff et al., 1999).
The appropriateness of a chapter on Shigella in a book devoted to pathogenic E. coli derives from the fact that EIEC cause dysentery that is clinically indistinguishable from that caused by members of the Shigella genus. In addition, an abundance of studies starting with the DNA–DNA hybridization work of Brenner et al. (1969) to multilocus enzyme electrophoresis (Pupo et al., 1997) and whole genome sequencing (Touchon et al., 2009; Sims and Kim, 2011) unequivocally establish that Shigella spp. are clones of E. coli. In this chapter, we will consider these pathogens together. Since the vast majority of the research on the pathogenesis of bacillary dysentery has been done on Shigella, we will focus on these organisms. No single review can be completely comprehensive. Therefore, the reader is encouraged to refer to several excellent recent reviews for additional information (Parsot, 2005; Schroeder and Hilbi, 2008).
Classification and biochemical characteristics
The Japanese microbiologist Shiga isolated an organism (Shigella dysenteriae) from the dysenteric stool of a stricken individual in 1898. Three more Shigella species (S. boydii, S. flexneri, and S. sonnei) were subsequently identified and grouped by serotype and metabolic activities. The four species of the genus Shigella are grouped serologically (41 serotypes) based on their somatic O-antigens: S. dysenteriae (group A), S. flexneri (group B), S. boydii (group C), and S. sonnei (group D). As members of the family Enterobacteriaceae, they are closely related to the salmonellae (Ochman et al., 1983). Shigella are non-motile, Gram-negative rods. Some important biochemical characteristics that distinguish these bacteria from other enterics are their inability to utilize citric acid as a sole carbon source and their inability to ferment lactose, although some strains of S. sonnei may ferment lactose slowly. They are oxidase-negative, do not produce H2S (except for S. flexneri 6 and S. boydii serotypes 13 and 14), and do not produce gas from glucose. Shigella spp. are inhibited by potassium cyanide and do not synthesize lysine decarboxylase (Ewing, 1986).
Based on the cumulative sequence evidence from 16S rRNA genes, multiple housekeeping genes, and whole genome comparisons, the pathogenic Shigella can be considered a subset of non-pathogenic E. coli (Cilia et al., 1996; Pupo et al., 1997; Shu et al., 2000; Jin et al., 2002). Although phylogenetically it would be more accurate to treat Shigella as pathotypes of E. coli, the members of the genus Shigella continue to be divided for historical and medical purposes into the four species or subgroups described above (Strockbine and Maurelli, 2005).
Enteroinvasive E. coli (EIEC) isolates that cause a diarrheal illness identical to Shigella dysentery were identified in the 1970s (DuPont et al., 1971; Tulloch et al., 1973). The pathogenic and biochemical properties that EIEC share with Shigella pose a problem for distinguishing these pathogens. For example, unlike normal flora E. coli, EIEC are non-motile and 70% of isolates are unable to ferment lactose (Silva et al., 1980; van den Beld and Reubsaet, 2012). These are also features of Shigella. More striking is the observation that strains of EIEC are almost universally negative for lysine decarboxylase (LDC) activity whereas almost 90% of normal flora E. coli are positive. In this respect, EIEC also resemble Shigella, which uniformly lack LDC activity. Some serotypes of EIEC even share identical O-antigens with Shigella (Sansonetti et al., 1985). By contrast, EIEC resemble E. coli in their ability to ferment xylose and to produce gas from glucose, both traits for which Shigella are negative (Silva et al., 1980).
It is now generally accepted that the present day strains of Shigella arose multiple times from as many as seven independent ancestral strains of E. coli (Pupo et al., 2000; Yang et al., 2005). EIEC probably evolved later than Shigella and from different E. coli ancestors (Lan et al., 2004). However, the common seminal event in the evolution of both groups of pathogens was the acquisition of a large plasmid that encodes the genes necessary for invasion of mammalian cells (see below) (Lan et al., 2001).
Evolution of Shigella species and EIEC
The EIEC were recognized as a heterogeneous group of pathogens that resembled Shigella in their pathogenic potential (and in certain metabolic traits) but were more closely related to E. coli. Early genetic studies demonstrated that essentially all the virulence factors required for expression of the invasive phenotype are encoded on a large plasmid present in all Shigella and EIEC isolates examined (Sansonetti et al., 1981, 1982b; Harris et al., 1982). Therefore, from the pathogenesis perspective, EIEC are closely related to Shigella. These observations led many investigators to propose, incorrectly, that the EIEC represented a missing link in the evolution from E. coli to Shigella.
Several studies have established that, like the EIEC, the four species of Shigella are so closely related to E. coli that they should all be included in a single species. The chromosomes of these organisms are largely co-linear and are more than 90% homologous (Brenner et al., 1969). Therefore, Shigella, like EIEC, are a group of pathogenic E. coli. In fact, several investigators using different approaches have established that the four species of Shigella evolved from separate E. coli strains (Pupo et al., 1997, 2000; Rolland et al., 1998). Seven different Shigella lineages have been identified through sequence analysis of multiple chromosomal loci. Bacteria expressing the Shigella phenotype were generated through horizontal transfer of the virulence plasmid from an unknown donor bacterium to commensal E. coli (Pupo et al., 2000). Thus, horizontal transfer of the Shigella virulence plasmid to commensal E. coli has occurred multiple times, each time giving rise to new Shigella clones. These findings suggest that traits unique to and shared by Shigella species and EIEC are the result of convergent evolution either through gain-of-function mutations (e.g. horizontal transfer of the virulence plasmid) or loss-of-function mutations (deletion of genes encoding traits expressed by ancestral E. coli strains). These insights provide a possible answer to lingering questions of the relationship of EIEC to Shigella and the evolution of these pathogens. The heterogeneous characteristics of EIEC are consistent with a polyphyletic history and reflect the non-clonal origins of these isolates. Moreover, these findings strongly suggest that the EIEC are not Shigella ancestors, or ‘missing links.’ Rather, the EIEC clones may be recently evolved pathogens that have not yet completely adapted to a pathogenic lifestyle and therefore have not fully developed the Shigella phenotype (Pupo et al., 2000). This proposal suggests that expression of the complete Shigella virulence phenotype requires not only the virulence plasmid but also additional modifications to the E. coli genome for optimal transmissibility, fitness in host tissues, and virulence.
Several recent studies have provided evidence of a new pathway of evolution, termed antagonistic pleiotrophy, that fine-tunes pathogen genomes for maximal fitness and virulence in host tissues. This pathway and the selective forces that drive its function can be observed in the transformation of commensal E. coli to virulent Shigella and EIEC pathogens. Following acquisition of the virulence plasmid and expression of its virulence genes, commensal E. coli gained access to new host tissues. However, such a newly evolved pathogen, which expresses the full complement of ancestral traits as well as virulence factors, may not be optimally suited for this new pathogenic lifestyle. Selective pressures encountered in the new environment (within colonocytes) are very different to those in the ancestral niche (lumen of the colon). In fact, some ancestral traits may interfere with the expression or function of factors required for survival within host tissues. Loci encoding these interfering factors are designated antivirulence genes. Thus, the newly evolved pathogen must inactivate ancestral antivirulence genes for optimal fitness and virulence. These modifications to the new pathogen’s genome are termed pathoadaptive mutations (Sokurenko et al., 1999; Maurelli, 2007).
The convergent evolution of the seven Shigella lineages and EIEC presents a unique opportunity for the identification and characterization of antivirulence genes and pathoadaptive mutations in addition to the study of pathogen evolution. Ancestral traits that interfere with virulence are lost from the newly evolved pathogen genome early on as the increased fitness of the adapted clones reinforces these beneficial mutations in the newly or recently evolved pathogen population. Therefore, some traits that are absent in all Shigella and EIEC, but commonly expressed in E. coli, are strong indicators of pathoadaptive mutations that have arisen by convergent evolution. Thus, evidence supporting this new pathogen evolution pathway was first provided in Shigella. Lysine decarboxylase (LDC) activity, which is encoded by the cadA gene, is expressed in over 85% of E. coli isolates. In contrast, no isolates of Shigella or EIEC express LDC activity (Silva et al., 1980). Lack of LDC activity in Shigella and EIEC is consistent with a pathoadaptive mutation. Experimental evidence of the antivirulence nature of the cadA gene was provided by the demonstration that the product of LDC activity, cadaverine, blocks the action of the virulence plasmid-encoded Shigella enterotoxin (Maurelli et al., 1998) and several other virulence phenotypes (McCormick et al., 1999). Sequence analysis of the cadA region of four Shigella lineages revealed novel genetic arrangements that are distinct in each strain examined (Day et al., 2001). Thus, the inactivation of cadA was accomplished by different mechanisms in each Shigella strain. Studies in EIEC showed that these strains have mutations in cadC, a transcriptional activator, which abolishes LDC activity (Casalino et al., 2003). These studies demonstrate that each newly evolved Shigella and EIEC clone increased virulence through the loss of an ancestral trait, LDC. Additional antivirulence loci that have been lost or inactivated in Shigella spp. and EIEC include nadA/nadB (Prunier et al., 2007) and speG (Barbagallo et al., 2011).
Molecular pathogenesis
Hallmarks of virulence
Shigella spp. and EIEC cause disease by overt invasion of epithelial cells in the large intestine. The clinical symptoms of dysentery can be directly attributed to the following hallmarks of virulence: induction of diarrhea, invasion of intestinal epithelial cells, multiplication inside these cells, spread from cell to cell, and stimulation of a strong host inflammatory response (Figure 7.1).
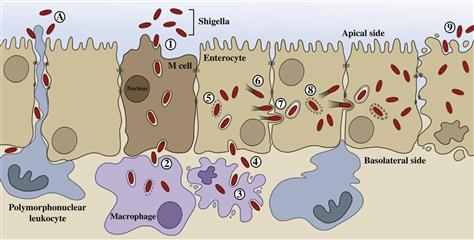
Shigella colonize the small intestine only transiently and cause little tissue damage (Rout et al., 1975). Production of enterotoxins by Shigella and EIEC in the small bowel probably results in the diarrhea that generally precedes onset of dysentery (Fasano et al., 1995; Nataro et al., 1995). It is believed that jejunal secretions elicited by these toxins facilitate passage of the bacteria through the small intestine and into the colon where they colonize and invade the epithelium.
Formal and his colleagues demonstrated the essential role of epithelial cell invasion in Shigella pathogenesis in a landmark study that employed both tissue culture assays to measure invasion and animal models (LaBree et al., 1964). They showed that spontaneous colonial variants of S. flexneri 2a that are unable to invade mammalian cells in tissue culture do not cause disease in monkeys. Further, they showed the presence of wild-type bacteria within epithelial cells of the large intestine in experimentally infected animals.
Gene transfer studies using E. coli K-12 donors and S. flexneri 2a recipients established the third hallmark of Shigella virulence. A S. flexneri 2a recipient that inherits the xyl-rha region of the E. coli K-12 chromosome retains the ability to invade epithelial cells but has a reduced ability to multiply within these cells (Falkow et al., 1963). This hybrid strain fails to cause a fatal infection in the opium-treated guinea pig model and is unable to cause disease when fed to rhesus monkeys (Formal et al., 1965). The high frequency of recombinants in these conjugation experiments also served to confirm the close genetic relatedness of E. coli and Shigella.
While it is necessary for Shigella to be able to multiply within the host epithelial cell after invasion, intracellular multiplication is not sufficient to cause disease. The bacterium must also be able to spread through the epithelial lining of the colon by cell-to-cell spread in a manner that does not require the bacterium to leave the intracellular environment and be re-exposed to the intestinal lumen (Figure 7.2). Mutants of Shigella that are competent for invasion and multiplication but are unable to spread between cells in this fashion have been isolated. These mutants established intracellular spread as the fourth hallmark of Shigella virulence and will be discussed in a later section.
Along with the ability to colonize and cause disease, an intrinsic part of any bacterium’s pathogenicity is the regulatory circuitry for controlling expression of virulence genes. Virulence in Shigella spp. and EIEC is regulated by temperature. After growth at 37°C, virulent strains of Shigella are able to invade mammalian cells but when cultivated at 30°C, they are non-invasive. This non-invasive phenotype is reversible by shifting the growth temperature to 37°C where the bacteria re-express their virulence factors (Maurelli et al., 1984). Similarly, the invasive phenotype of EIEC is also temperature regulated (Small and Falkow, 1988). Temperature regulation of virulence gene expression is a characteristic of other human pathogens such as pathogenic E. coli, Salmonella typhimurium, Bordetella pertussis, Yersinia spp., and Listeria monocytogenes. Regulation of gene expression in response to environmental temperature is a useful strategy for bacteria. This strategy permits Shigella and EIEC to trigger gene expression by sensing the ambient temperature of the mammalian host (i.e. 37°C) thereby economizing energy that would otherwise be expended on the synthesis of virulence products when the bacteria are outside the host. The system also permits the bacteria to coordinately regulate expression of multiple unlinked genes that are required for the full virulence phenotype.
Cell biology
Shigellosis, or bacillary dysentery, is an acute inflammatory disease of the colonic mucosa that results from invasion and cell-to-cell spread of the bacteria. Infection occurs via the oral route by ingestion of the bacteria in contaminated food or water. Shigella and EIEC display a remarkable ability to survive the acidity of the stomach (Gorden and Small, 1993; Jennison and Verma, 2007). After passing through the stomach, the bacteria transit through the small intestine to reach the colon where infection is established. Studies in several different animal models demonstrate that Shigella gain access to the colonic subepithelium and establish an infection through M cells. These specialized cells do not produce microvilli and are not covered with mucus or glycocalyx. They sample intestinal antigens which are transcytosed unmodified to underlying lymphoid follicles. In the subepithelium, the bacteria are rapidly ingested by resident macrophages. However, rather than being killed, Shigella escape the phagosome and induce caspase-1 activation and pyroptosis of the infected macrophage (Zychlinsky et al., 1992; Suzuki et al., 2005). Death of the phagocyte has dual effects as the bacteria are released unharmed along with the pro-inflammatory cytokine IL-1β that initiates an inflammatory cascade (Zychlinsky and Sansonetti, 1997). Following release from the macrophage, Shigella penetrate colonic epithelial cells through the basolateral surface via interaction with α5-β1 integrins and CD44 (Watarai et al., 1996). As in the macrophage, internalized bacteria rapidly escape the endosomal vacuole (Ray et al., 2010) and spread to adjacent cells. In contrast to the fate of the macrophage, Shigella actively block induction of apoptosis in epithelial cells (Faherty and Maurelli, 2009). Infection of the colonocytes induces the release of still more pro-inflammatory cytokines, IL-6 and IL-8 (Sansonetti et al., 1999). The high levels of pro-inflammatory cytokines produced by epithelial cells and macrophages in response to Shigella infection signal the massive recruitment of polymorphonuclear neutrophils (PMNs), the only cells that are able to kill Shigella. Thus a paradox of Shigella–host interaction exists: how can the shigellae be successful pathogens (able to infect new hosts) when the organisms are readily killed and eventually cleared by the massive innate host immune response? One possible answer has recently been proposed.
The severe signs and symptoms of shigellosis are due to extensive tissue destruction by the intense inflammatory response. The major effectors of this damage are PMNs. Inhibition of PMN recruitment to the site of infection using anti-IL-8 antibodies significantly reduces the severity of symptoms (Sansonetti et al., 1999). PMN degranulation mediates tissue destruction and destabilizes tight junctions between colonocytes (Perdomo et al., 1994b). Shigella present in the intestinal lumen exploit this window of opportunity to gain access to the subepithelium where the pathogens can begin another round of infection by invasion of colonocytes through the basolateral surface (Perdomo et al., 1994a). Intracellular bacteria are likely protected from PMN killing. Thus, the initial focus of infection is amplified through successive rounds of infection made possible by the activity of PMNs. Amplification of the infection increases the number of Shigella in the infected host and increases the likelihood of successful transmission to another host. Consistent with this model, recruitment of PMNs across the epithelium to the apical side (which faces the intestinal lumen) requires virulent, invasive Shigella (McCormick et al., 1998). Recruitment of PMNs further requires that the bacteria interact with the basolateral membrane of the epithelial cells, where IL-8 is secreted. Bacteria present on the apical side do not efficiently invade colonocytes nor do they recruit PMNs. Shigella residing within cells (or in the subepithelium) mediate PMN recruitment across the epithelium to the apical surface in model monolayers in part by inducing the secretion of the potent PMN chemoattractant, hepoxilin A3 (Mumy et al., 2008). Secretion of hepoxilin A3 from the apical surface of the epithelium creates a chemotactic gradient that guides PMNs from the submucosa to the apical surface of the epithelium. Collectively, these observations suggest that Shigella plays dangerous games with effectors of the host innate immune response by first inducing, then exploiting, the intense inflammatory response to ensure its own survival and evolutionary success (passage to another host).
Virulence genes
Plasmid-encoded
Given the complexity of the interactions between host and pathogen, it is not surprising that the virulence of Shigella and EIEC is multigenic, involving both chromosomal and plasmid-encoded genes. Another landmark study on the pathogenicity of Shigella was the demonstration of the indispensable role for a large plasmid in invasion. A 180 kilobase pair (kb) plasmid in S. sonnei and a 220 kb plasmid in S. flexneri were first shown to be required for invasion (Sansonetti et al., 1981, 1982b). Other Shigella spp. as well as strains of EIEC also contain large plasmids that are functionally interchangeable and share significant degrees of DNA homology with the plasmid described in S. flexneri (Harris et al., 1982; Sansonetti et al., 1982a). These plasmids can be classified into two distinct but closely related forms based on sequence analysis (Lan et al., 2001). Analysis of the complete DNA sequence of the 220 kb plasmid in S. flexneri 5a reveals a complex history (Buchrieser et al., 2000; Venkatesan et al., 2001). This plasmid is essentially a genetic mosaic composed of blocks of genes from different origins as well as the remnants of four different ancestral plasmids. For instance, the genes required for the invasive phenotype have a G+C content of 31–37%. This value is significantly lower than the composition of loci required for intercellular spread, and the composition of these loci is significantly different from loci encoding a surface protease. These genes are interspersed with close to 100 insertion sequences comprising one-third of the virulence plasmid. Evidence exists of active transposition, rearrangements, and deletions in the plasmid mediated by homologous recombination between these elements. These events likely led to the size and organizational differences observed between the virulence plasmids of different Shigella species and lineages.
A 37 kb region of the invasion plasmid of S. flexneri 2a contains all of the genes necessary to permit the bacteria to penetrate into tissue culture cells. This DNA segment was identified as the minimal region of the virulence plasmid necessary to allow a plasmid-cured derivative of S. flexneri (and E. coli K-12) to invade tissue culture cells (Maurelli et al., 1985). The 34 virulence genes encoded in this region are organized into three large operons (ipa, mxi, and spa) that are transcribed in opposite directions. The essential role of these genes in expression of the invasive Shigella phenotype and virulence has been demonstrated using mutation analyses. The organization of these loci in large transcription units suggests that the genes are co-regulated and encode factors that function together in a multicomponent virulence (invasion) system. The mxi and spa operons encode subunits of a type III secretion system (T3SS; see Chapter 14) and the ipa operon encodes proteins (effectors; see Chapter 15) secreted by this system. T3SS are widely used by animal and plant pathogens (enterohemorrhagic and enteropathogenic E. coli, Salmonella spp., Yersinia spp., Pseudomonas spp., Erwinia caratovara, and Xanthomonas campestris, to name a few) for delivery of virulence factors to the surface or interior of host cells (Galan and Wolf-Watz, 2006). These secreted effectors of virulence allow the pathogens to reprogram host cells to serve the pathogen’s particular needs. The T3SS delivery machines are conserved structurally, functionally, and genetically and share several core characteristics.
T3SS genes
The products of the ipa genes are actively secreted into the extracellular medium even though they contain no signal sequence for recognition by the usual general secretory pathway of Gram-negative bacteria. Ipa secretion is mediated by a T3SS composed of gene products from the mxi/spa loci (Schroeder and Hilbi, 2008). A characteristic feature of T3SS is a contact-dependent secretion mechanism. Delivery of effectors to their targets (surface and cytoplasmic) occurs only after outer membrane elements of the T3SS complex contact cholesterol in the host cell membrane (Hayward et al., 2005). This feature is observed in the Shigella T3SS as the Ipa proteins are deposited on the host cell surface after contact with host cells (Menard et al., 1994a; Watarai et al., 1995).
The structural elements of the Shigella T3SS are encoded in two adjacent operons on the virulence plasmid. The mxi (membrane expression of invasion plasmid antigens) genes encode several lipoproteins (MxiJ and MxiM), a transmembrane protein (MxiA), and proteins containing signal sequences (MxiD, MxiJ, and MxiM) (Schroeder and Hilbi, 2008). MxiH is the major subunit of the secretion needle (Blocker et al., 2001). The spa (surface presentation of Ipa antigens) genes encode additional components of the Shigella T3SS. The cytoplasmic protein Spa32 controls the switch for T3SS substrate specificity and thus regulates needle length (Magdalena et al., 2002; Tamano et al., 2002). Assembly of the T3SS needle and subsequent effector secretion requires Spa47 which is associated with the cytoplasmic face of the secretion apparatus and has sequence similarities with ATPases of the flagellar assembly machinery of other bacteria (Vankatesan et al., 1992; Tamano et al., 2000). Spa47 likely is the energy-generating component of the secretion apparatus.
Invasion plasmid antigens
The genes comprising the ipaBCDA (invasion plasmid antigens) cluster encode the immunodominant antigens of Shigella and EIEC that are detected with sera from convalescent patients and experimentally challenged monkeys (Hale et al., 1985; Oaks et al., 1986). ipaBCD are absolutely required for invasion of mammalian cells and carry out post-invasion roles as secreted effectors (Menard et al., 1993). An ipaA mutant is still invasive but has a 10-fold reduced ability compared to wild-type (Tran et al., 1997). This operon also encodes a molecular chaperone, IpgC (invasion plasmid gene), which binds IpaB and IpaC in the bacterial cytosol prior to secretion and prevents their interaction and proteolytic degradation (Menard et al., 1994b). Following secretion, IpaB and IpaC form a complex that binds to molecules such as α5-β1 integrins and CD44 that are present on the basolateral surface of host cells (Watarai et al., 1996; Skoudy et al., 2000). After binding, the Ipa complex inserts into the host cell membrane and causes a massive recruitment of host cell actin and other cytoskeletal factors beneath the site of attachment. This exploitation of the host cell cytoskeleton is observed with IpaBC-coated beads or whole Shigella bacteria and leads to the formation of localized membrane projections that encompass and internalize either beads or bacteria in roughly 5 minutes (Menard et al., 1996). Purified IpaC induces cytoskeletal reorganization, including formation of filopodia and lamellipodial extensions on permeabilized cells (Tran et al., 1999). Inside the host cell, IpaA interacts with vinculin to promote F-actin depolymerizaton. Through this action, IpaA may promote formation of a pseudo-focal adhesion complex that organizes the actin assembly complex beneath the bacterium and directs optimal uptake of the pathogen (Tran et al., 1997; Bourdet-Sicard et al., 1999).
IpaB serves several other roles in Shigella virulence that may be accomplished by separate domains of the protein. IpaB is required for escape of Shigella from both the phagosomal (following uptake by macrophages) and endosomal (following uptake by epithelial cells) compartments (High et al., 1992). This activity requires formation of a hydrophobic N-terminal alpha-helix and is linked to the ability of IpaB to insert into host cell membranes and lyse erythrocytes. In addition, IpaB binds to pro-caspase 1 in vitro and in vivo and triggers hydrolysis of the cysteine protease (Zychlinsky et al., 1994). In macrophages, this activity leads to secretion of IL-1β (which initiates inflammation) and induction of pyroptosis of the phagocyte.
The needle tip protein, IpaD, forms a pentameric ring at the end of the T3SS needle and prevents secretion of the effector proteins (Espina et al., 2006). IpaD also acts as an environmental sensor that triggers recruitment of IpaB to the needle tip upon exposure to bile salts (Olive et al., 2007). Upon contact with the cholesterol-rich plasma membrane of the mammalian cell, IpaC is recruited to the needle tip where it associates with IpaB to form a translocon pore within the host cell membrane (Epler et al., 2009). The IpaB/IpaC translocon is the conduit through which effectors pass directly from the bacterium into the host cell cytoplasm.
IpgB1 plays a critical role in invasion. It acts as a mimic of the host small GTPase RhoG at the host plasma membrane, activates Rac1, and stimulates membrane ruffling through the ELMO-Dock180 pathway (Ohya et al., 2005; Handa et al., 2007). IpgB2, a homolog of IpgB1, mimics the GTP-bound form of RhoA and triggers formation of actin stress fibers (Alto et al., 2006). IpgD displays phosphatidylinositol 4-phosphatase activity and induces morphological changes in the infected cell including membrane blebbing and ruffling at the entry site (Niebuhr et al., 2002).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


