- Normal cells in culture divide a finite number of times before they experience growth arrest and stop dividing – a largely stable and irreversible state termed “replicative senescence.”
- Senescence results from the cumulative negative effect of repeated cycles of replication on important structures at the ends of chromosomes (telomeres).
- Progressive shortening and loss of function of the telomeres result in chromosome ends being treated as damaged DNA with resultant activation of DNA damage responses (DDRs).
- The activity of a key enzyme, telomerase, can prevent telomere attrition.
- Cancer cells need telomerase to maintain functional telomeres and to divide indefinitely, and this has opened the possibility of novel therapeutic approaches based on using telomerase as a target.
- Telomerase is reactivated in over 90% of all types of human tumor.
- Senescence may result from mechanisms independent of telomerase activity and telomere attrition, particularly in response to environmental factors and DNA damage that share common downstream signaling involving the p53–p21CIP1 and RB–p16INK4a pathways, which collaborate to stop cellular proliferation.
- The RB and p53 tumor suppressor pathways are required to maintain stable growth arrest in senescent cells in vitro. In fact, at least in cultured cells, senescence may be reversible by inactivating these pathways. Therefore, the possibility should be considered that senescent cells could still contribute to cancer at a future date by re-entering the cell cycle.
- Oncogene-induced senescence has been shown to restrain transformation in human cancer cells and cancer progression in animal models in vivo, and together with apoptosis is a key innate restraint on the potential of oncogene deregulation to give rise to cancer.
- Stem cells are a likely cell of origin for many cancers, as they already exhibit many of the properties required of cancer cells, including longevity, lesser degrees of differentiation, and advanced replicative potential. Cancer originating by acquired mutations in stem cells would appear to have a head start on those originating in more differentiated cells. One caveat is that deregulation of some oncogenes can confer stem cell–like properties on previously differentiated cells.
- Irrespective of their origin, cancers appear to contain a particularly malign side population of cells potentially responsible for metastases, recurrence, and resistance to treatments. These cells are often referred to as “cancer stem cells” due to their similar phenotype to normal stem cells, to which their relationship is still hotly debated.
Introduction
Billions of bilious barbecued blue blistering barnacles!
Hergé
This chapter will cover some of the most important and intriguing concepts in cancer biology. Prominent among these are the effect of senescence as a barrier to tumorigenesis and how this may be overcome and the role of stem cells and cancer stem cells in the initiation and progression of cancers. We will discuss several processes such as telomere maintenance, RB and p53 signaling, and various factors involved in regulating stemness and epithelial–mesenchymal transition (EMT). Inevitably there will be overlap with other chapters, in particular Chapters 7 and 12.
Cells are regarded as senescent when they lose replicative potential but remain living and metabolically active. Senescence is a tumor suppressor mechanism which may serve as a related but alternative option to apoptosis in preventing the propagation of a cancer-causing mutation. Many stimuli that can promote apoptosis can also result in senescence, including deregulated oncogene activity, DNA damage, and telomere attrition. Moreover, there is considerable overlap between the signaling pathways regulating apoptosis and senescence, particularly those involving p53. However, Rb and Skp2 appear to be more specifically involved in growth arrest and senescence as compared to apoptosis. Intriguingly, some senescence-specific signaling pathways appear to suppress apoptosis, suggesting that at least in some cases apoptosis may be a last-resort option when growth arrest or senescence fails to allow damage repair or appears to the cell to be a futile effort in the face of serious DNA damage or other stress. Recent studies, as has been the case with many other areas of cell biology, have revealed important roles for microRNAs (miRNAs) in regulating senescence. Senescence is associated with alterations in chromatin as well as specific secretory patterns, knowledge which has been employed to identify senescent cells in tissues and cell cultures. The importance of epigenetic mechanisms is underscored by the central role of the senescence-related histone methyltransferase Suv39h1 in mediating senescence induced by TGF-β and by deregulated oncogene activation.
Most human cells in culture can reproduce only a limited number of times before they lose the ability to divide and become what is called “senescent.” This process has been associated with various aspects of longevity determination and aging, and it also may play an important role in preventing cancer.
Seminal work by Hermann Muller and Barbara McClintock identified that the ends of chromosomes are capped by structures, which serve to prevent chromosome fusions, the telomeres. As the mechanics of DNA replication were described, it was identified that DNA polymerase, the enzyme responsible for DNA replication, could not fully synthesize the 3′ end of linear DNA. This was described by James Watson as the end-replication problem. At about the same time in Russia, Alexey Olovnikov proposed that the end-replication problem would result in telomere shortening with each round of replication and that this mechanism could account for replicative senescence, which was compatible with studies by Leonard Hayflick and colleagues that control of replicative senescence was situated in the nucleus. Olovnikov’s model is supported by numerous studies which show that telomere shortening is a major contributor to replicative senescence and that telomere length may act as a “mitotic clock” to determine the number of cell divisions allowed for a given cell (see Box 9.1, “Hayflick limit”).
Telomerase is a reverse-transcriptase enzyme that elongates the telomeres, thus counteracting the normal telomere attrition occurring every time DNA is replicated. Telomerase has two components, an RNA component and a catalytic subunit, and is strongly implicated in the avoidance of senescence by “immortal” tumor cell lines. Studies have also confirmed that expression of the catalytic subunit of human telomerase (hTERT) in various cell types avoids replicative senescence by maintaining telomere length. Telomerase is not the only mechanism capable of elongating the telomeres, although the mechanisms underlying this alternative lengthening of telomeres (ALT) remain unknown. Whatever the mechanisms, stabilizing telomeres is a prerequisite for immortality and likely for tumor development, normally by telomerase. In contrast, telomerase inhibition can induce senescence in cancer cells, but if cells with shortened telomeres manage to divide, this may also under these circumstances be a strong supporter of cancer progression due to the increased rate of subsequent chromosomal aberrations (Fig. 9.1). Most, if not all, human somatic tissues have no detectable telomerase activity; in the bone marrow, hematopoietic cells express telomerase. Telomerase activity is higher in primitive progenitor cells and then downregulated during proliferation and differentiation. Telomerase activity has been detected in several normal human somatic proliferating cells, for instance skin and colorectal tissues. Expression of hTERT appears to be regulated by the proto-oncoprotein c-MYC, thus providing a potential link between cell-cycle progression and telomere maintenance. As is so often the case, increased study does not always result in increased simplicity. Telomerase reverse transcriptase (TERT) has been implicated as a regulator of cell proliferation independently of its actions at the telomere, and may influence key proliferative signaling pathways, such as WNT, directly by interaction with the protein BRG1.
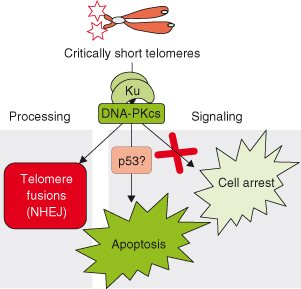
Figure 9.1 Critically short telomeres lead to end-to-end telomere fusions and loss of cell viability (apoptosis and/or cell-cycle arrest). The nonhomologous end-joining (NHEJ) complex DNA-PK has been involved both in mediating telomere fusions and in apoptosis triggered by critically short telomeres. In particular, these consequences of telomere dusfunction are rescued in DNA-PKcs-deficient and Ku86-deficient backgrounds. However, cell-cycle arrest due to short telomeres is not rescued by the absence of DNA-PKcs and Ku86 proteins. We have suggested that p53 may be downstream of DNA-PK in signaling apoptosis due to short telomeres.
Although telomere length regulates replicative senescence, it is not the sole regulator of cell senescence. Telomeres are not linear, but form duplex loops called “t-loops” (Fig. 9.2), the formation of which is dependent on telomeric repeat-binding factors TRF1 and TRF2, and may be crucial for stabilizing telomere caps. Capping may protect the telomeres from being recognized as DNA damage and may be as important as telomere length in avoiding telomere dysfunction and preventing apoptosis and senescence.
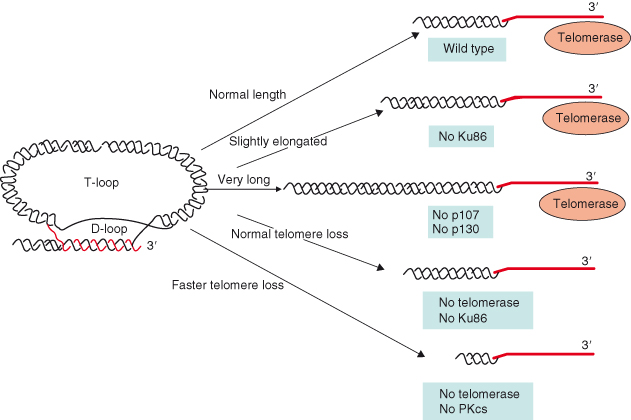
Figure 9.2 Different activities modulate telomere length, such as the telomere-binding proteins TRF1 and TRF2. More recently, activities involved in DNA repair (Ku86 and DNA-PKcs) and in cell-cycle regulation (p107 and p130) have also been shown to have a direct impact in regulating telomere length. Ku86 is a negative regulator of telomerase, while DNA-PKcs cooperates with telomerase in maintaining telomere length. Simultaneous absence of p107 and p130 results in a fast elongation of telomeres in the absence of changes in telomerase activity.
In a recent publication, an intriguing link between telomere crisis and the progression of a form of breast cancer has been identified. It was found using an in vitro model that ductal hyperplasia may be associated with a critical shortening of telomeres in rapidly dividing cells resulting in crisis. Importantly, although the majority of such cells undergo apoptosis or presumably senescence, thus avoiding further risk of progression to cancer, a small number of cells may escape these usual consequences of chromosome instability. Instead rare cells may reactivate telomerase, thus preventing further telomere attrition and allowing such cells to progress to ductal carcinoma in situ and presumably thence to invasive cancer. Not surprisingly, the rate of genetic aberrations and chromosomal re-arrangements was highest during the crisis period. It is speculated that such a transition through a telomere crisis may be a crucial event in the development of most breast carcinomas.
Senescence can occur in fibroblasts in the absence of cell division and short telomeres. Although one hypothesis is that telomere dysfunction occurs in confluent cells despite a lack of telomere shortening, it is also apparent that viral genes such as SV40T-antigen, adenoviral E1A and E1B, and the human papillomavirus E6 and E7 genes can also immortalize cells. The E1B and E6 proteins bind and inactivate the tumor suppressor protein p53, while E1A and E7 bind and inactivate the retinoblastoma protein (RB, also spelled pRb). Immortalization depends on inactivation of p53, RB, or their pathways.
At present, it is accepted that the p53 and RB pathways are largely responsible for inducing senescence. Immortalization with viral proteins or oncogenes results in an extended lifespan after which cells enter a stage called “crisis,” where cells proliferate but the rate of apoptotic cells gradually increases and thus cell numbers eventually diminish. Since both the p53 and RB–p16INK4a pathways are inactive and chromosomal instability and fusions are abundant, crisis is thought to emerge due to extremely short telomeres. Occasionally, immortal cells emerge from crisis with stabilized telomeres, normally involving telomerase activation. In a sense, crisis can be seen as the ultimate consequence of telomere dysfunction. Whatever changes occur during telomere dysfunction, the mechanisms triggering growth arrest appear to involve the recognition of dysfunctional telomeres as DNA damage and that the p53 and RB–p16INK4a pathways collaborate to stop cellular proliferation.
Importantly, even in some normal tissues a cell subpopulation exists, the stem cells, which already avoids many of the restraints posed by these processes. Not surprisingly, therefore, it is now widely assumed that many cancers may originate from these cells rather than more differentiated ones, because stem cells have a head start in the evolutionary race to malignancy. Closely allied to the role of stem cells as the origin of cancers is the role of cells within most cancers which have some stem cell properties (wherever they may have come from originally). These so-called cancer stem cells (CSCs) are now one of the most intensively studied areas in all of cancer biology. In brief, the CSC hypothesis states that cancers are maintained by a subpopulation of cells which manifest self-renewal (the motor for cancer) and differentiation (these cells generate heterogeneous progeny that proceed in some way along one or more differentiation pathways forming the bulk of the tumor).
Senescence
Getting older is no problem. You just have to live long enough.
Groucho Marx
To summarize what has been discussed, cellular senescence represents a stable state in which cells manifest proliferative arrest; it is a potentially desirable consequence of some anticancer treatments and conversely one of the barriers that must be overcome for malignant transformation to proceed. Senescence depends on alterations in chromatin and chromatin-dependent tumor suppression. However, it is worth noting that senescence may actually have evolved as a means of expediting repair. In senescence, terminal differentiation is induced by a variety of stimuli including alterations of telomere length and structure, some forms of DNA damage (e.g. oxidative stress), and activation of certain oncogenes.
Senescence differs from other physiologic forms of cell-cycle arrest such as quiescence in that it is largely irreversible, barring the inactivation of p53 and/or RB, and it is associated with distinctive phenotypic alterations such as cellular flattening, the expression of senescence-associated β-galactosidase activity, and the establishment of an unusual form of heterochromatin known as senescence-associated heterochromatic foci (SAHFs). These SAHFs may provide a chromatin buffer to silence proliferation-associated genes.
Senescence requires activation of the p16INK4a–RB and the p53–p21CIP1 pathways. The CKIs, p16INK4a and p21CIP1 (inactive in many cancers), inhibit the activities of CDK4 and CDK6 and of CDK2, CDK4, and CDK6, respectively, thus preventing cell-cycle progression (Chapters 4 and 7). Exogenous expression of p16INK4a induces senescence in immortal cells lacking functional p53, and conversely cells may be immortalized by disruption of both p16INK4a and p53. RB is needed for growth suppression mediated by p16INK4a suggesting that it acts upstream of RB in regulating senescence. Telomere dysfunction can activate p16INK4a and also p53. The p53 pathway is an important mediator of senescence that is induced by dysfunctional telomeres, in part by triggering DNA damage responses (Chapter 10). The cell-cycle can be blocked by p21CIP1 independently of p16INK4a (Fig. 9.3).
Figure 9.3 (a) Growth arrest can be induced by both p53–p21 and p16INK4a–Rb-dependent mechanisms. (b) Linkage between signaling regulating replication, apoptosis, and senescence. Activation of RAS and MYC results in potential engagement of both replication and growth as well as of apoptosis and possibly senescence. If either MYC or RAS levels are excessive (as might occur during oncogenesis) or other pro-apoptotic signals are received, then the balance may be tipped away from replication. RAS can promote senescence through either p16INK4a or ARF, which activate the RB or p53 pathways respectively. Senescence may also result from telomere erosion or dysfunction, which may activate DNA damage responses that are partly the same as those also activated by DNA DSBs. Functional telomeres must normally prevent DNA ends from being recognized and processed as DSBs. MYC can inhibit growth arrest or senescence by inhibiting p21CIP1 and inducing telomerase reverse transcriptase (TERT), which prevents telomere erosion and may also activate replication by other means. Although MYC may activate the apoptotic pathway (e.g. via ARF), RAS is able to suppress apoptosis by activating the PI3K pathway and, subsequently, AKT. It can readily be appreciated how oncogenic MYC and RAS may conspire in oncogenesis. The combination of RAS and MYC acting together provides a potential means of avoiding apoptotic and senescence mechanisms activated by either acting individually. Moreover, it can also be appreciated how inactivating mutations in RB or p53 (or their pathways involving p19Arf, p16INK4a, p21CIP1, etc.) may contribute to tumorigenesis by enabling the cancer cell to avoid senescence, apoptosis, or both.
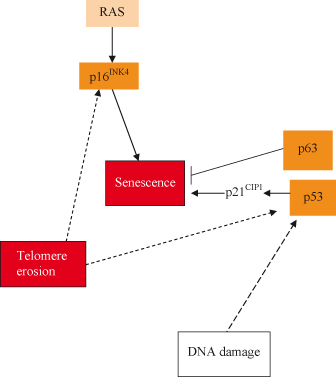
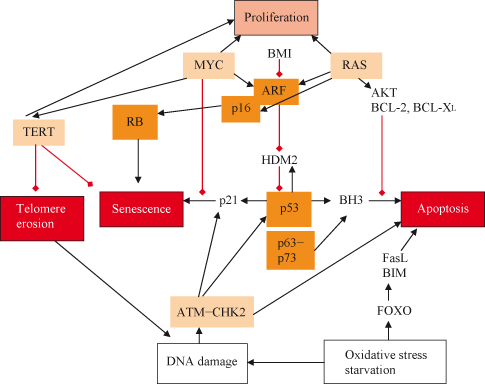
Stabilization of p53 occurs in response to DNA damage and also to inappropriate activation of oncogenes such as c-MYC via induction of ARF (p19ARF in the mouse), which can bind MDM2, thereby inhibiting the destruction of p53 (Chapter 7). Either p53–p21CIP1 or p16INK4a can promote RB hypophosphorylation and initiate senescence. Some senescence-inducing stimuli (e.g. activation of the RAS oncogene) appear to induce both pathways, while others (e.g. DNA damage) appear to preferentially activate one or the other. Recent studies confirm that diverse potentially cancer-promoting genes can trigger senescence in vivo, including oncogenic RAS and BRAF, which appear to induce p16INK4a via the ERK pathway and activation of AKT or inactivation of PTEN, which activate the ARF–p53–p21CIP1 pathway.
Numerous pathways can affect p16INK4a and influence senescence. Thus, Hedgehog signaling and the GLI2 transcription factor can prevent senescence. Conversely, signaling via TGF-β can promote senescence. A recent study has shown that cells derived from mice with knockout of both p16INK4a and p21CIP1 do not senesce in serial culture and can override Ras-induced senescence, which is not the case if only one or the other is inactivated. The authors also found these mice to be prone to skin cancer and suggest that loss of p16INK4a promoted benign tumors which progressed to cancer under the influence of loss of p21CIP1. The actual mechanisms involved will likely vary between different tumors but share a common final denominator, namely, prevention of RB phosphorylation and therefore inhibition of the G0–G1 to S transition. Intriguingly, RB may also promote senescence by interacting with various factors that can result in histone methylation and the formation of transcriptionally inactive heterochromatin. This may stably silence S-phase genes in senescent cells. This raises some questions over the use of drugs that can reverse such epigenetic changes (Chapter 11). Recently it has been shown in a mouse model of lymphoma that inactivation of a Suv39h1, a key histone methyltransferase often associated with RB, prevented senescence and fostered cancer progression. Many drugs targeting epigenetic modification (inhibitors of histone deacetylases and DNA methyltransferases) with the aim of reversing epigenetic silencing of tumor suppressors (Chapter 11) could conceivable also reverse or prevent senescence. I think that by now the reader will be well aware of the pervasive presence of functionally important miRNAs in essentially all areas of biology. Recent studies show that the RB pathway can upregulate miR-29 and miR-30; miRNA families are upregulated during induced and replicative senescence, and by inhibiting B-Myb expression contribute to cell-cycle arrest.
During advancing age p16INK4a accumulates, providing a molecular link between aging and senescence; p16INK4a inhibits CDK4 (Chapter 4), which is required for proliferation in adult cells. Recent studies show that defective replication and regeneration in mouse pancreatic β cells of adult transgenic mice with increasing age are mediated by increasing p16INK4a. Interestingly, this effect is tissue specific and is not observed in the exocrine pancreatic cells of the same animals; moreover, only p16INK4a of the CKIs tested was increased in an age-dependent manner. Finally, mice lacking p16INK4a have improved regeneration and proliferation of β cells, raising the possibility that the effects of aging on cell growth could potentially be reversed therapeutically, though the effect this might have on cancer incidence would be a concern.
Much progress has been made on unraveling the complex crosstalk and other signaling pathways that affect cellular senescence. Thus, recent studies have implicated the activation of the NF–kB pathway and a variety of downstream gene targets as positive regulators of senescence. The tumor suppressor promyelocytic leukemia protein (PML), discussed in Chapter 11, is also an important regulator of senescence and terminal differentiation, in part by a well-described activation of p53 and through a more recently described PML–RB–E2F pathway that acts by localizing E2F transcription factors bound to their promoters and RB to PML nuclear bodies.
Senescent cells with irreparable DNA damage contain nuclear foci that contain DNA damage response (DDR) proteins, which persist and involve PML bodies in contrast to the transient complexes seen in cells with repairable DNA lesions that do not. Campisi and coworkers have recently described what they refer to as “DNA segments with chromatin alterations reinforcing senescence” (DNA-SCARS), in which DNA synthesis is absent. These contain histone H2AX, activated CHK2, and p53 and do not contain DNA repair proteins such as RPA and RAD51. Although not required for DNA-SCARS, p53 and RB are needed to trigger the senescence growth arrest.
A recent study has employed genome-wide expression profiling with genetic complementation in order to assess genes that were differentially expressed when conditionally immortalized human fibroblasts were induced to senesce by activating the p16–RB and p53–p21 tumor suppressor pathways. The expression of more than 1700 genes, of which 50% have been previously shown to be upregulated in cancers, was reversed when senescence was bypassed, leading the authors to note that cancers appear to devote considerable efforts to overcoming senescence. The study also suggested a central role of the NF-κB signaling pathway in maintaining senescence.
Cellular Senescence, Immortality, and Cancer
I don’t believe one grows older. I think that what happens early on in life is that at a certain age one stands still and stagnates.
T.S. Eliot
In 1961, Leonard Hayflick and Paul Moorhead made the unexpected discovery that human cells derived from embryonic tissues can divide only a finite number of times in culture (Hayflick and Moorhead, 1961 – and see Box 9.1 on the Hayflick limit). Hayflick and Moorhead worked with a connective tissue cell, the fibroblast, but similar findings have been observed for other cell types including skin keratinocytes, endothelial cells, and lymphocytes. Importantly, senescence also occurs in cells derived from embryonic tissues and in cells taken from mice and other animals. Early results suggested a relation between the number of “population doublings” that cells undergo in culture, and the longevity of the species from which the cells were derived. For example, cells from the Galapagos tortoise, which can live longer than a century, divide about 110 times, while mouse cells divide roughly 15 times. If cells are taken from individuals with premature aging syndromes, such as Werner’s syndrome (the gene responsible for Werner’s syndrome, WRN, is a member of the RecQ subfamily of DNA helicases), they exhibit far fewer doublings than normal cells.
Some cells never reach replicative senescence and are said to be “immortal.” These cells include embryonic germ cells and most cell lines derived from tumors. Replicative senescence in human fibroblasts is characterized by growth arrest (i.e. the cells stop dividing, are larger and morphologically heterogeneous, and are more sensitive to cell–cell contact inhibition). Senescent cells growth arrest at the G1–S transition of the cell cycle, which is in general irreversible (inactivation of p53 and RB pathways notwithstanding), and cells remain refractory to growth factors even though senescent cells can remain metabolically active for long periods of time.
Replicative senescence may limit the growth potential, but not necessarily survival, of a dividing cell as a consequence of telomere attrition. Senescent cells exhibit a particular phenotype when arrested in the G1 phase of the cell cycle. They appear flattened and enlarged with increased cytoplasmic granularity, enhanced activity of senescence-associated lysosomal hydrolase β-galactosidase when assessed at an acidic pH. In 1995, Judith Campisi and her team discovered that the enzyme β-galactosidase is abnormal in senescent cells; senescence-associated β-galactosidase activity becomes active at a higher pH (pH 6 rather than 4). Early reports showed that lysosomes increase in the number and size in senescent cells, and the increased autophagy with aging in culture may be associated with an increase of lysosomal mass. While refractory to mitogenic stimuli, senescent cells remain viable and metabolically active and possess a typical transcriptional profile that distinguishes them from quiescent cells. At the protein level, numerous regulators of cell-cycle progression, checkpoint control, and cellular integrity such as p53 or p16INK4a have been found to be induced in response to various pro-senescent stimuli. Although the molecular mechanisms underlying the senescent phenotype remain largely unknown, there is increasing evidence that the formation of heterochromatin in the vicinity of promoters that control gene expression related to cell-cycle progression might be implicated in the maintenance of an irreversible growth arrest. This accumulation of SAHFs in senescent cells can act as a chromatin buffer able to prevent the transcriptional activation of proliferation-associated genes. Recent work from the laboratory of Scott Lowe has shown that SAHFs in senescent fibroblasts contain High-Mobility Group A (HMGA) proteins, which can cooperate with the p16INK4a both in the formation of SAHF and also in proliferative arrest. Interestingly, HMGA-mediated growth arrest is negated by coexpression of HDM2 and CDK4 oncogenes, which are often coamplified with HMGA2 in human cancers, and presumably limit activity of the Rb and p53 tumor suppressor pathways.
Normal human cells are diploid, yet with each passage in culture, the proportion of polyploid cells increases. Mutations in mitochondrial DNA also increase with age. Senescent cells are less able to express heat shock proteins, which may also increase their vulnerability to death triggers. The expression of multiple genes is altered during cellular aging. Normally, cell culture conditions include 20% oxygen, and these were the conditions initially used by Hayflick and Moorhead. However, if human fibroblasts are cultured at reduced oxygen (3%) closer to physiological conditions, they achieve a further 20 doublings. In contrast, different types of human cells cultured above 20% oxygen survive fewer doublings. Importantly, the same effect is not observed in tumor cell lines.
Cell senescence can be found without telomere shortening in vivo. One view is that replicative senescence is not a major factor in vivo and that cellular senescence instead depends on stress factors that trigger the RB and p53 tumor suppressor pathways. From a simple mathematical perspective, the existence of replicative senescence under normal physiological conditions in vivo has been questioned. The reasoning is along these lines: assuming human fibroblasts endure 50 population doublings, 250 would seem more than enough cells to last several lifetimes under normal conditions. However, in cancer the issue has recently been somewhat clarified. First, deregulated expression of oncogenes seems to trigger cell cycles which in some way are detected by the cell as aberrant (cancer cell cycles). These cancer cell cycles trigger DNA damage responses, likely independently of telomere attrition, which culminate in growth arrest or apoptosis. These responses variously involve activation of the ARF–p53 or p16INK4a–RB pathway. Whether the detection of DNA damage or the activation of DNA damage responses is essential for this remains to be shown.
Stem cells participate in tissue homeostasis by replacing differentiated cells that have undergone cell death or been lost (e.g. shed from the skin surface). Human stem cells express the enzyme telomerase suggesting that these actively dividing cells could avoid telomere attrition by replacing telomeric repeats. However, somatic stem cells can show senescence, and telomere-independent mechanisms of cellular senescence are increasingly being described.
The purpose of cellular senescence is unclear, but it may act as an anticancer barrier. Loss-of-function mutations in WRN lead to genomic instability, an elevated cancer risk, and premature cellular senescence. Interestingly, WRN is normally upregulated by c-MYC, suggesting that WRN may act downstream of c-MYC to prevent cellular senescence during tumorigenesis (Chapter 6). Recent studies suggest that in addition to WRN, two other MYC-regulated proteins, CDK2 and telomerase, may also mitigate MYC-induced senescence and contribute to the oncogene cooperation seen between MYC and other senescence-inducing oncogenes such as RAS. Interestingly, unlike WRN, loss of CDK2 does not contribute to oncogene stress and apoptosis.
It is also worth noting, as was discussed in Chapter 6, that the binding of MYC with MIZ1 is essential for the repression of various CKIs, required for efficient promotion of the G1–S transition. It also appears that in some tumors related to deregulated MYC, such as lymphomas, TGF-β expression is greatly increased and in the absence of MYC activation (or if this is inactivated) will drive senescence associated with accumulation of trimethylated histone H3 at Lys 9 (H3K9triMe). Whilst MYC is active, however, H3K9triMe is suppressed. The authors propose that TGF-β expression may contribute to addiction to deregulated MYC in these cancers, as the high levels of expression would induce senescence once MYC was targeted or inactivated.
Epidemiological studies have indicated for a long time that cancer rates accelerate dramatically after a person reaches age 50, an observation usually attributed to an accumulation of harmful genetic mutations. However, Judith Campisi and others have suggested that the accumulation of cells with a senescent phenotype (persisting in certain tissues after undergoing changes in morphology, behavior, and function) might also contribute to cancer. Certainly, even in animal models it has been shown that telomere attrition may not invariably be an anticancer mechanism. Thus, if cells are able to avoid replicative senescence (e.g. by losing p53) despite telomere shortening, then they are at increased risk of undergoing major chromosomal aberrations during cell division, which may actually support cancer progression.
Senescent Cells May Promote Cancer Behaviors in Other Cells: the Dangers of Old Stroma
The young are always ready to give to those who are older than themselves the full benefits of their inexperience.
Oscar Wilde
Senescent cells may secrete many different molecules, some of which have been shown by Campisi and colleagues to have a “field effect” that could encourage malignant behavior in nearby cells in a paracrine fashion. This has resulted in speculation that cellular senescence evolved as a cancer suppression mechanism at a time when the life expectancy for humans was far shorter than it is today. Given that most people now can expect a much greater life expectancy (at least in developed countries), senescence may be an example of “antagonistic pleiotropy.” Namely, a trait selected to optimize fitness early in life turns out to have unselected deleterious effects later on. In fact, such mechanisms, including fetal programming, have been proposed as contributors to many of the more prevalent diseases of modern life including not only cancer but also obesity, diabetes, and atherosclerotic diseases. One version of this is the so-called Barker hypothesis, whereby exposure of a fetus to malnutrition during ontogeny may in some way foster the birth of a child “programmed” to successfully cope with malnutrition through adult life. The subsequent exposure of such an “adapted” individual to an unexpectedly energy-rich diet of burgers, pizzas, and sugar-containing drinks, for which that individual is now maladapted, may then result in disease, albeit usually at an older age.
These digressions aside, cellular senescence is an effective barrier to neoplastic transformation as it removes risky cells from the cell division cycle. If, however, senescent cells can secrete factors that stimulate the proliferation of nonsenescent cells – potentially including premalignant cells – then we have an intriguing situation in which a tumor suppressor response becomes a pro-tumorigenic one, termed “antagonistic pleiotropy.” In a study from the laboratory of Judith Campisi, premalignant mammary epithelial cells exposed to senescent human fibroblasts in mice irreversibly lose differentiation and undergo malignant transformation. One of the secreted factors from senescent cells that may contribute to this behavior is the matrix-degrading enzyme MMP3. Thus, senescent cells in stromal tissues may contribute to age-related tumorigenesis by secreting factors that can alter the differentiation state of other cell types, such as epithelial cells in the evolving tumor. Intriguingly, MMP3 produced by stromal cells has recently been shown to influence various processes in a cancer (or would-be cancer) cell. In addition to fostering invasion and metastases at later stages in tumor development, MMPs may exert an early influence on cell transformation and cancer development. MMP3 can activate a member of the RAC family (RAC1b) promoting the formation of reactive oxygen species (ROS) with resultant DNA damage and genomic instability. Moreover, MMP3 can also induce EMT, a major factor in cancer formation and spread, possibly by cleaving E-cadherin and triggering signaling via the WNT–β-catenin pathway.
A further recent study has suggested that stromal-derived TGF-β (in this study, released from macrophages engulfing apoptotic lymphoma cells) can promote senescence in MYC-induced lymphomas.
Premature Senescence, Reversibility, and Cancer
Whatever poet, orator, or sage may say of it, old age is still old age.
Henry Wadsworth Longfellow
Premature senescence recapitulates the cellular and molecular features of replicative senescence (see also Chapter 6 on oncogene-induced senescence). Extrinsic factors such as anticancer agents, γ-irradiation, and UV light have been shown to induce premature senescence as a DNA damage–mediated cellular stress response. DNA lesions are sensed and transduced via protein complexes involved in DNA maintenance and repair including the ataxia telangiectasia mutated kinase (ATM), among others (Chapter 10). Such kinases directly or indirectly phosphorylate the p53 protein at certain residues, which can then participate in apoptosis or in the induction of DNA damage–mediated senescence. The CKI p16INK4a is implicated in both response to DNA damage and control of stress-induced senescence, although how p16INK4a induces permanent G1 arrest is not known. In fact, p16INK4a is essential for the maintenance of cellular senescence. The p21CIP1 protein acts as a molecular switch that triggers telomere-initiated senescence. Interestingly, the mechanisms by which dysfunctional telomeres lead to p21CIP1 activation are similar, but not identical, to cellular responses to DNA damage. The p16INK4a protein acts independently from telomeres (discussed further in this chapter).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


