High-Yield Terms to Learn
Sedation Reduction of anxiety Addiction The state of response to a drug whereby the drug taker feels compelled to use the drug and suffers anxiety when separated from it Anesthesia Loss of consciousness associated with absence of response to pain Anxiolytic A drug that reduces anxiety, a sedative Dependence The state of response to a drug whereby removal of the drug evokes unpleasant, possibly life-threatening symptoms, often the opposite of the drug’s effects Hypnosis Induction of sleep REM sleep Phase of sleep associated with rapid eye movements; most dreaming takes place during REM sleep Sedation Reduction of anxiety Tolerance Reduction in drug effect requiring an increase in dosage to maintain the same response
Pharmacokinetics
Absorption and Distribution
Most sedative-hypnotic drugs are lipid-soluble and are absorbed well from the gastrointestinal tract, with good distribution to the brain. Drugs with the highest lipid solubility (eg, thiopental ) enter the CNS rapidly and can be used as induction agents in anesthesia. The CNS effects of thiopental are terminated by rapid redistribution of the drug from brain to other highly perfused tissues, including skeletal muscle. Other drugs with a rapid onset of CNS action include eszopiclone, zaleplon, and zolpidem.
Metabolism and Excretion
Sedative-hypnotics are metabolized before elimination from the body, mainly by hepatic enzymes. Metabolic rates and pathways vary among different drugs. Many benzodiazepines are converted initially to active metabolites with long half-lives. After several days of therapy with some drugs (eg, diazepam, flurazepam), accumulation of active metabolites can lead to excessive sedation. Lorazepam and oxazepam undergo extrahepatic conjugation and do not form active metabolites. With the exception of phenobarbital, which is excreted partly unchanged in the urine, the barbiturates are extensively metabolized. Chloral hydrate is oxidized to trichloroethanol, an active metabolite. Rapid metabolism by liver enzymes is responsible for the short duration of action of zolpidem. A biphasic release form of zolpidem extends its plasma half-life. Zaleplon undergoes even more rapid hepatic metabolism by aldehyde oxidase and cytochrome P450. Eszopiclone is also metabolized by cytochrome P450 with a half-life of 6 h. The duration of CNS actions of sedative-hypnotic drugs ranges from just a few hours (eg, zaleplon < zolpidem = triazolam = eszopiclone < chloral hydrate) to more than 30 h (eg, chlordiazepoxide, clorazepate, diazepam, phenobarbital).
Mechanisms of Action
No single mechanism of action for sedative-hypnotics has been identified, and the different chemical subgroups may have different actions. Certain drugs (eg, benzodiazepines) facilitate neuronal membrane inhibition by actions at specific receptors.
Benzodiazepines
Receptors for benzodiazepines (BZ receptors) are present in many brain regions, including the thalamus, limbic structures, and the cerebral cortex. The BZ receptors form part of a GABA A receptor-chloride ion channel macromolecular complex, a pentameric structure assembled from 5 subunits each with 4 transmembrane domains. A major isoform of the GABAA receptor consists of 2  1, 2
1, 2 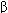 2, and 1
2, and 1  2 subunits. In this isoform, the binding site for benzodiazepines is between an
2 subunits. In this isoform, the binding site for benzodiazepines is between an  1 and the
1 and the  2 subunit. However, benzodiazepines also bind to other GABAA receptor isoforms that contain
2 subunit. However, benzodiazepines also bind to other GABAA receptor isoforms that contain  2,
2,  3, and
3, and  5 subunits. Binding of benzodiazepines facilitates the inhibitory actions of GABA, which are exerted through increased chloride ion conductance (Figure 22-1).
5 subunits. Binding of benzodiazepines facilitates the inhibitory actions of GABA, which are exerted through increased chloride ion conductance (Figure 22-1).
FIGURE 22-1

A model of the GABA A receptor-chloride ion channel macromolecular complex. A hetero-oligomeric glycoprotein, the complex consists of 5 or more membrane-spanning subunits. Multiple forms of  ,
, 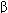 , and
, and  subunits are arranged in various pentameric combinations so that GABAA receptors exhibit molecular heterogeneity. GABA appears to interact at two sites between
subunits are arranged in various pentameric combinations so that GABAA receptors exhibit molecular heterogeneity. GABA appears to interact at two sites between  and
and 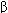 sububits, triggering chloride channel opening with resulting membrane hyperpolarization. Binding of benzodiazepines and the newer hypnotic drugs such as zolpidem occurs at a single site between
sububits, triggering chloride channel opening with resulting membrane hyperpolarization. Binding of benzodiazepines and the newer hypnotic drugs such as zolpidem occurs at a single site between  and
and  subunits, facilitating the process of chloride ion channel opening. The benzodiazepine antagonist flumazenil also binds at this site and can reverse the hypnotic effects of zolpidem. Note that these binding sites are distinct from those of the barbiturates.
subunits, facilitating the process of chloride ion channel opening. The benzodiazepine antagonist flumazenil also binds at this site and can reverse the hypnotic effects of zolpidem. Note that these binding sites are distinct from those of the barbiturates.
(Reproduced, with permission, from Katzung BG, editor: Basic & Clinical Pharmacology, 11th ed. McGraw-Hill, 2009: Fig. 22-6.)
Benzodiazepines increase the frequency of GABA-mediated chloride ion channel opening. Flumazenil reverses the CNS effects of benzodiazepines and is classified as an antagonist at BZ receptors. Certain  -carbolines have a high affinity for BZ receptors and can elicit anxiogenic and convulsant effects. These drugs are classified as inverse agonists.
-carbolines have a high affinity for BZ receptors and can elicit anxiogenic and convulsant effects. These drugs are classified as inverse agonists.
Barbiturates
Barbiturates depress neuronal activity in the midbrain reticular formation, facilitating and prolonging the inhibitory effects of GABA and glycine. Barbiturates also bind to multiple isoforms of the GABAA receptor but at different sites from those with which benzodiazepines interact. Their actions are not antagonized by flumazenil. Barbiturates increase the duration of GABA-mediated chloride ion channel opening. They may also block the excitatory transmitter glutamic acid, and, at high concentration, sodium channels.
Other Drugs
The hypnotics zolpidem , zaleplon, and eszopiclone
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


