The calculated parameter for the Vd has no direct physical equivalent; therefore, it is usually denoted the apparent Vd. A drug that is completely retained in the plasma compartment (Figure 3-1) will have a Vd equal to the plasma volume (about 4% of body weight). The Vd of drugs that are normally bound to plasma proteins such as albumin can be altered by liver disease (through reduced protein synthesis) and kidney disease (through urinary protein loss). On the other hand, if a drug is avidly bound in peripheral tissues, the drug’s concentration in plasma may drop to very low values even though the total amount in the body is large. As a result, the Vd may greatly exceed the total physical volume of the body. For example, 50,000 liters is the average Vd for the drug quinacrine in persons whose average physical body volume is 70 liters.
FIGURE 3-1
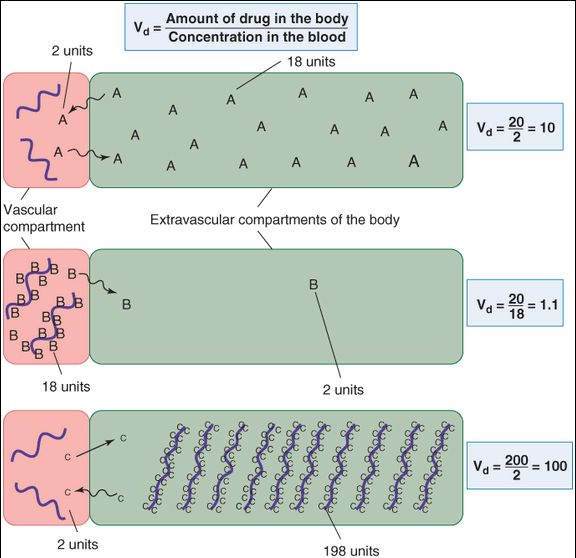
Effect of drug binding on volume of distribution. Drug A diffuses freely between the 2 compartments and does not bind to macromolecules (heavy wavy lines) in the vascular or the extravascular compartments of the hypothetical organism in the diagram. With 20 units of the drug in the body, the steady-state distribution leaves a blood concentration of 2 units. Drug B, on the other hand, binds avidly to proteins in the blood. At equilibrium, only 2 units of the total are present in the extravascular volume, leaving 18 units still in the blood. In each case, the total amount of drug in the body is the same (20 units), but the apparent volumes of distribution are very different. Drug C is avidly bound to molecules in peripheral tissues, so that a larger total dose (200 units) is required to achieve measurable plasma concentrations. At equilibrium, 198 units are found in the peripheral tissues and only 2 units in the plasma, so that the calculated volume of distribution is greater than the physical volume of the system.
Clearance
Clearance (CL) relates the rate of elimination to the plasma concentration:

For a drug eliminated with first-order kinetics, clearance is a constant; that is, the ratio of rate of elimination to plasma concentration is the same regardless of plasma concentration (Figure 3-2). The magnitudes of clearance for different drugs range from a small percentage of the blood flow to a maximum of the total blood flow to the organs of elimination. Clearance depends on the drug and the condition of the organs of elimination in the patient. The clearance of a particular drug by an individual organ is equivalent to the extraction capability of that organ for that drug times the rate of delivery of drug to the organ. Thus, the clearance of a drug that is very effectively extracted by an organ (ie, the blood is completely cleared of the drug as it passes through the organ) is often flow-limited. For such a drug, the total clearance from the body is a function of blood flow through the eliminating organ and is limited by the blood flow to that organ. In this situation, other conditions—disease or other drugs that change blood flow—may have more dramatic effects on clearance than disease of the organ of elimination. Note that for drugs eliminated with zero-order kinetics (see Figure 1-3, right), clearance is not constant.
FIGURE 3-2
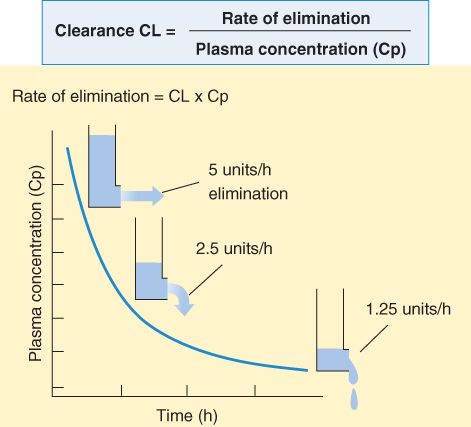
The clearance of the great majority of drugs is relatively constant over a broad range of plasma concentrations. Since elimination rate is equal to clearance times plasma concentration, the elimination rate will be rapid at first and slow as the concentration decreases.
Skill Keeper 1: Zero-Order Elimination
(See Chapter 1)
Most drugs in clinical use obey the first-order kinetics rule described in the text. Can you name 3 important drugs that do not? The Skill Keeper Answer appears at the end of the chapter.
Half-Life
Half-life (t 1/2) is a derived parameter, completely determined by Vd and CL. Like clearance, half-life is a constant for drugs that follow first-order kinetics. Half-life can be determined graphically from a plot of the blood level versus time (eg, Figure 1-4), or from the following relationship:
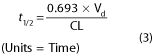
One must know both primary variables (Vd and CL) to predict changes in half-life. Disease, age, and other variables usually alter the clearance of a drug much more than they alter its Vd. The half-life of a drug may not change, however, despite a decreased clearance if the Vd decreases at the same time. This occurs, for example, when lidocaine is administered to patients with heart failure. The half-life determines the rate at which blood concentration rises during a constant infusion and falls after administration is stopped (Figure 3-3). The effect of a drug at 87-90% of its steady-state concentration is clinically indistinguishable from the steady-state effect; thus, 3-4 half-lives of dosing at a constant rate are considered adequate to produce the effect to be expected at steady state with a specified rate of chronic dosing.
FIGURE 3-3
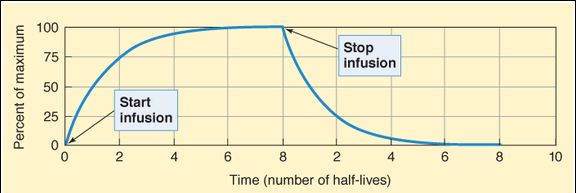
Plasma concentration (plotted as percent of maximum) of a drug given by constant intravenous infusion for 8 half-lives and then stopped. The concentration rises smoothly with time and always reaches 50% of steady state after 1 half-life, 75% after 2 half-lives, 87.5% after 3 half-lives, and so on. The decline in concentration after stopping drug administration follows the same type of curve: 50% is left after 1 half-life, 25% after 2 half-lives, and so on. The asymptotic approach to steady state on both increasing and decreasing limbs of the curve is characteristic of drugs that have first-order kinetics.
Bioavailability
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


