CHAPTER 6 Research Strategies
R esearch in cell biology aims to discover how cells work at the molecular level. Powerful tools are now available to achieve this goal. To understand how these methods contribute to the broad effort to explain cellular function, this chapter begins with a brief account of the synthetic approach used in cell biology. This strategy is based on the premise that one can understand a complex cellular process by reducing the system to its constituent parts and characterizing their properties. This approach, also called reductionism, has dominated cell biology research since the middle of the 20th century and has succeeded time after time. For example, most of what is understood about protein synthesis has come from isolating and characterizing ribosomes, messenger RNAs (mRNAs), transfer RNAs (tRNAs), and accessory factors. In this and many other cases, proof of function has been established by reconstituting a process from isolated parts of the molecular machine and verifying these conclusions with genetic experiments.
This agenda is complete for remarkably few biological processes. Bacterial chemotaxis is one example (see Figs. 27-12 and 27-13). Often, much is known about some aspects of a process, such as a partial list of participating molecules, the localization of these molecules in a cell, or a test for function by removing the genes for one or more molecules from an experimental organism. Rarely is enough information available about molecular concentrations and reaction rates to formulate a mathematical model of the process to verify that the system actually works as anticipated. Thus, much work remains to be done.
Box 6-1 is a guide for locating descriptions of methods used throughout this book. This chapter begins with imaging, one extremely valuable method for studying cells. Microscopy of live and fixed cells often provides initial hypotheses about the mechanisms of cellular process. It is also a valuable adjunct to genetic analysis and testing mechanisms. The chapter then covers a selection of other methods that are used for cell biology research.
BOX 6-1 Guide to Experimental Methods Discussed throughout This Book
| Method | Pages |
|---|---|
| Light microscopy | 86–90 |
| Electron microscopy | 90–92 |
| Gene and protein identification by classical genetics | 94–95 |
| Gene and protein identification by genomics and reverse genetics | 95–96 |
| Protein purification | 96–99 |
| Gel electrophoresis | 97 |
| Column chromatography | 98 |
| Organelle purification | 96 |
| Isolation of genes and cDNAs (PCR, cloning) | 99–102 |
| Molecular structure (hydrodynamics, X-ray crystallography, NMR) | 102 |
| Identification of binding partners by biochemistry | 102–103 |
| Identification of binding partners by genetics and genomics | 103–105 |
| Reaction rates and affinities | 105 |
| Microscopic localization of proteins and nucleic acids | 105–106 |
| Physiological tests of function by genetics | 106–107 |
Imaging
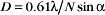
Light Microscopy
A half dozen optical tricks are used to produce contrast in light micrographs of biological specimens (Table 6-1 and Fig. 6-1). These are called wide-field methods, as a broad beam of illuminating light is focused on the specimen by a condenser lens.
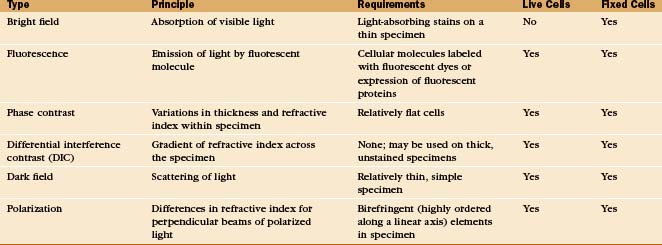
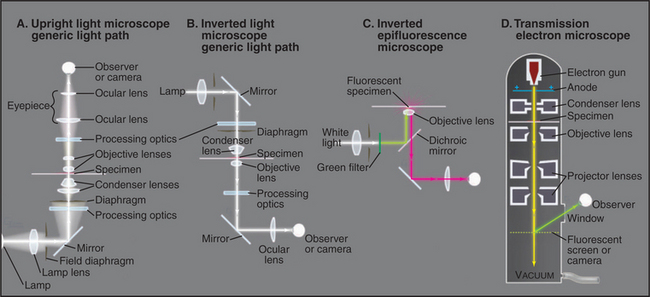
The classic light microscopic method is bright field, whereby the specimen is illuminated with pure white light. Most cells absorb very little visible light and thus show little contrast with bright-field illumination (Fig. 6-2A). For this reason, staining is used to increase light absorption and contrast. Because staining makes it difficult to see through thick tissues, specimens must also be relatively thin, about 1 mm for critical work. Slides for histologic and pathological study are produced by fixing cells with cross-linking chemicals, embedding them in paraffin or plastic, making sections with a microtome (a device that cuts a series of thin slices from the surface of a specimen), and staining with a variety of dyes (for examples, see Figs. 28-2, 28-5, 28-6, 28-7, 29-3, 29-8, 32-1, and 32-2). Alternatively, thin slices may be taken from frozen tissue and then stained. In either case, the cells are killed by fixation or sectioning prior to observation.
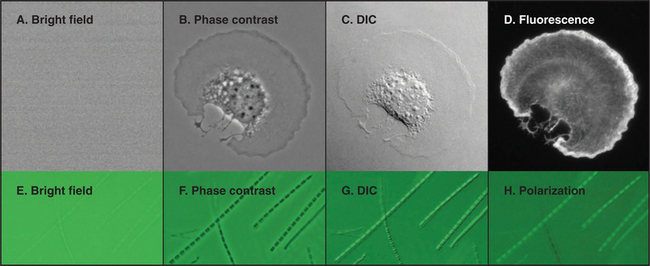
Figure 6-2 comparison of methods to produce contrast. a–d, Micrographs of a spread mouse 3T3 cell grown in tissue culture on a microscope slide, then fixed and stained with rhodamine-phalloidin, a fluorescent peptide that binds actin filaments. Contrast methods include bright field (A), phase contrast (B), differential interference contrast (C), and fluorescence (D). E–H, Micrographs of myofibrils isolated from skeletal muscle. Contrast methods include bright field (E), phase contrast (F), differential interference contrast (G), and polarization (H). The A-bands, consisting of parallel thick filaments of myosin (see Fig. 39-3), appear as dark bands with phase contrast and are birefringent (either bright or dark, depending on the orientation) with polarization.
(A–D, Courtesy of R. Mahaffy, Yale University, New Haven, Connecticut.)
Observations of live cells require other methods to produce contrast. In every case, these methods are also useful for fixed cells. Phase-contrast microscopy generates contrast by interference between light scattered by the specimen and a slightly delayed reference beam of light. Small variations in either thickness or refractive index (speed of light) can be detected, even within specimens that absorb little or no light (Fig. 6-2 B). Differential interference contrast (DIC) produces an image that looks as though it is illuminated by an oblique shaft of light (Fig. 6-2 C). What actually happens is that two nearby beams interfere with each other, producing contrast in proportion to local differences (gradient) in the refractive index across the specimen. Thus, a vesicle with a high refractive index (slow speed of light) in cytoplasm will appear light on one side (where the refractive index is increasing with respect to the cytoplasm) and dark on the other (where the refractive index is decreasing).
Fluorescence microscopy requires a fluorescent dye or protein in the specimen. Remarkable sensitivity makes fluorescence microscopy a powerful tool. Under favorable conditions, single fluorescent dyes or fluorescent protein molecules can be imaged. When a fluorescent molecule absorbs a photon of light, an electron is excited into a higher state. Nanoseconds later, a longer-wavelength (lower-energy) photon is emitted when the electron falls back to its ground state. For example, the fluorescent dye rhodamine absorbs green light (shorter wavelength) and emits red light (longer wavelength). Fluorescence microscopes use filters and special dichroic mirrors that reflect short wavelengths of light used to illuminate and excite fluorescent specimens but transmit the longer-wavelength emitted fluorescent light into the imaging system (camera). Strategically placed emission filters remove the exciting light reflected by the specimen so that only the fluorescent regions of the specimen appear bright. To provide fluorescence, a purified lipid, protein, or nucleic acid can be labeled with a fluorescent dye and injected into a live cell, where it will seek its natural location (see Figs. 37-6 and 38-9). Molecules labeled with a fluorescent dye can also be used to locate a target in a fixed and permeabilized cell. A powerful version of this strategy uses antibodies, proteins produced by the immune system (see Fig. 28-9), to react with specific molecular targets. Antibodies are tagged with fluorescent dyes and used to localize molecules in fixed cells by fluorescence microscopy (Fig. 6-3 E). This is called immunofluorescence. Another strategy is to label an oligonucleotide with a fluorescent dye to probe for nucleic acids with complementary sequences in fixed cells (see Fig. 13-15). Yet another approach is to localize individual structures, such as actin filaments, with a fluorescent dye attached to a small peptide that binds tightly to these filaments (Fig. 6-2 D).
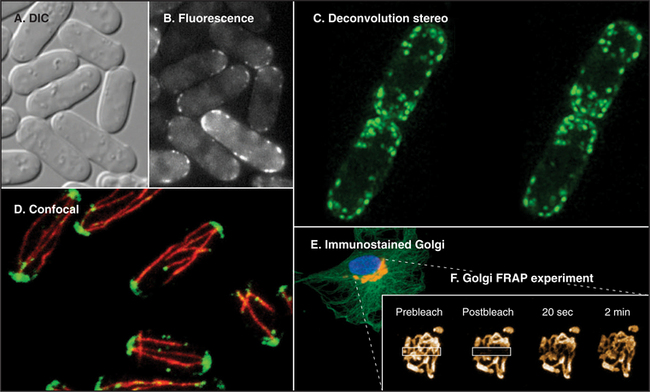
The discovery of proteins whose amino acid sequence renders them naturally fluorescent, such as green fluorescent protein (GFP) from jellyfish, made fluorescence microscopy immensely valuable for observation of individual proteins in live cells. Typically, DNA-encoding GFP is joined to one end of the coding sequence for a cellular protein and introduced into cells, which then synthesize a fusion protein consisting of GFP linked to the protein of interest. GFP fluorescence marks the fusion protein wherever it goes in the cell and can be quantified to determine how many labeled molecules reside in a particular cellular location (Fig. 6-3). Ideally, the coding sequence for GFP fusion protein is inserted into the genome of the test cell in place of the wild type gene, and the fusion protein is shown to function normally by genetic or biochemical experiments. Where this is difficult or impossible (e.g., in most studies of metazoan cells), the GFP fusion protein can be produced from exogenous DNA or RNA introduced into the cell. Mutations in GFP can change its fluorescence properties, providing probes in a range of colors and with differing sensitivities to distinct biochemical parameters in the cell, such as pH, Ca2+ concentration, and kinase activity. When attached to different protein types, these probes allow two or more protein species to be visualized simultaneously in the same cell and can serve as “biosensors” to measure changes in the intracellular environment and in a protein’s behavior/interactions.
Dark-field microscopy and polarization microscopy have specialized uses in biology. In dark-field microscopy, the specimen is illuminated at an oblique angle so that only light scattered by the specimen is collected by the objective lens. Recall how easy it is to detect tiny dust particles in a beam of light in a dark room. The contrast is so great that single microtubules stand out brightly from the dark background. However, for the images to be interpretable, the specimen must be very simple, much simpler than a cell. A dark-field image of something as complicated as cytoplasm is very confusing, owing to multiple overlapping objects that scatter light.
Like dark-field microscopy, polarization microscopy produces a bright image on a dark background. When a specimen is viewed between two crossed polarizing filters, only light whose polarization state is modified by the specimen will pass through the second polarizer to the image. Polarization microscopy relies on a specimen’s crystalline order, or birefringence, to provide contrast. Birefringent specimens, such as filaments in striated muscle (Fig. 6-2 H) or microtubules in a mitotic spindle, are aligned enough that polarized light, oriented so that it vibrates along the length of the polymers, passes through more slowly than does light vibrating perpendicular to the polymers (much as a knife cuts through meat faster with the grain than across it). Most cells do not have sufficient birefringence to produce a useful image with a conventional polarization microscope. New methods are making this approach more applicable for future work.
Computer processing can greatly enhance contrast and remove optical artifacts from images. For example, computer-enhanced DIC can image single microtubules (see Fig. 34-7). New methods of image processing can even improve detection beyond the classic limit determined by the wavelength of light (about 0.2 mm with green light). A processing method called deconvolution produces clear fluorescence images of thick specimens by using an iterative computer process to restore light that is blurred out of focus to its proper focal plane. Starting with a stack of blurry images taken at different focal planes all the way through the specimen using a traditional wide-field microscope, this method produces a remarkably detailed three-dimensional image in sharp focus throughout (Fig. 6-3 C).
Confocal microscopy also produces thin optical sections of fluorescent specimens. Rather than illuminating with a wide beam of light, this method uses a point of laser light sharply focused in all three directions: x, y, and z. The point of light is scanned across the specimen in a raster pattern (checkerboard pattern, like the electron beam in a TV) to excite fluorescent molecules. Light emitted at each consecutive point in the specimen passes through a pinhole placed next to the detector to remove any light that does not come directly from each focal point. A computer reassembles the image from the fluorescence at each point in this checkerboard of fluorescence signals (Fig. 6-3 D; see also Figs. 14-17 and 14-18 Figs. 13-12, 14-2, and 44-23). A series of confocal images taken at different planes of focus can be used for three-dimensional reconstructions.
Electron Microscopy
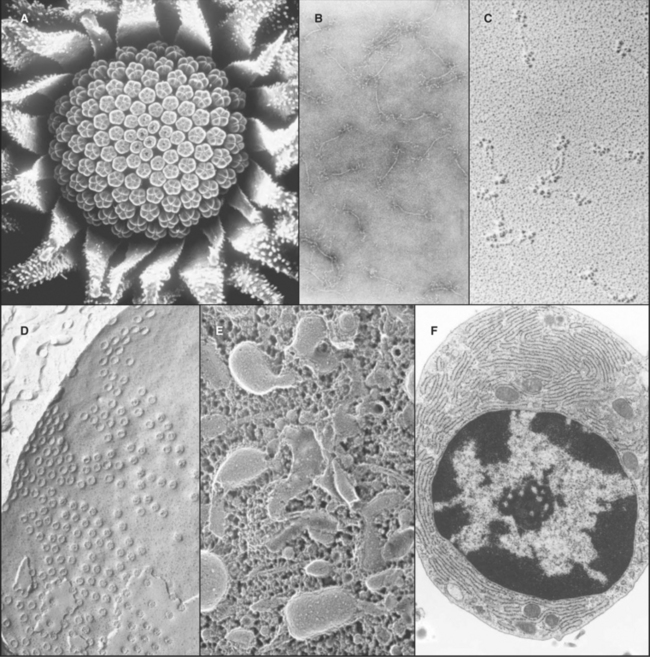
A transmission electron microscope (Fig. 6-1 D) can resolve points below 0.3 nm, but the practical resolution is usually limited by damage to the specimens from the electron beam and the methods used to prepare specimens. Historically, the most common method used to prepare cells for electron microscopy was to fix the specimen with chemicals, embed it in plastic, cut the specimen into thin sections, and stain the sections with heavy metals (Fig. 6-4 F). With this technique, the resolution is limited to about 3 nm, but that is sufficient to bridge the gap between light microscopy and molecular structures. During the heyday of electron microscopy in cell biology, between 1950 and 1970, thin sections revealed most of what is known about the organization of organelles in cells.
The highest resolution is attained with regular specimens, such as two-dimensional protein crystals rapidly frozen and viewed while embedded in a thin film of vitreous (i.e., amorphous, noncrystalline) ice (see Fig. 5-11A). This is called cryoelectron microscopy be-cause the stage holding the frozen specimen is cooled to liquid nitrogen temperature. Electron micrographs and electron diffraction of frozen crystals have produced structures of bacteriorhodopsin (see Fig. 7-8), aquaporin water channels (see Fig. 10-15), and tubulin (see Fig. 34-4) at resolutions of 3 to 4 nm. Computational image processing methods are used to calculate the three-dimensional structure of proteins in these regular specimens. These methods are similar to those used to calculate electron density maps from X-ray diffraction patterns (see Fig. 3-10). Although the resolution is limited and data collection is tedious in electron crystallography, electron microscopic images have the advantage of containing the phase information that is often difficult to ascertain with X-ray diffraction.
Electron microscopy is valuable for studying protein polymers and other large macromolecular specimens at less-than-atomic resolution. Diverse methods are used to prepare specimens and impart contrast. One way is to freeze filaments or macromolecular assemblies in vitreous ice, as described earlier (see Figs. 34-7 and 36-4A). A second is negative staining, whereby specimens are dried from aqueous solutions of heavy metal salts (Fig. 6-4 B). A shell of dense stain encases particles on the surface of a thin film of carbon and can preserve structural details at a resolution of about 1 nm. Alternatively, macromolecules dried on a smooth surface can be shadowed with a thin coat of metal evaporated from an electrode (Fig. 6-4 C). A variation of this approach that improves preservation is to freeze specimens rapidly, evaporate the ice surrounding the molecules, and then apply a coat of platinum (see Figs. 30-4 and 34-11).
Computer image processing of micrographs of certain types of structures can yield an average three-dimensional reconstruction of a molecular structure. Particles with helical symmetry, such as actin filaments (see Fig. 33-7) and microtubules (see Fig. 34-5), are analyzed by an image-processing method called deconvolution to reconstruct the three-dimensional structure. Single particles may also be reconstructed by first classifying images of thousands of randomly oriented particles into categories corresponding to different views. Then, an average three-dimensional structure is calculated computationally from this ensemble. One example is the Sec61p translocon associated with a ribosome (see Fig. 20-6). More recently, computing advances have led to the development of electron microscope tomography, in which many pictures are taken of a relatively thick specimen from different angles (by tilting the speci-men inside the microscope). Superimposition blurs each picture, but when they are merged together into a three-dimensional map, structures as complex as entire cells can be visualized at a resolution of a few nanometers.
Cells and tissues can also be frozen rapidly and prepared for electron microscopy without chemical fixation. In the freeze-fracture method, the frozen specimen is cleaved to expose the inside of the cells, and exposed surfaces are rotary-shadowed with a thin coat of platinum. This surface coat is then viewed by using a transmission electron microscope (Fig. 6-4 D). Frequently, the cleavage plane splits lipid bilayers in half to reveal proteins embedded in the plane of the membrane. If some of the frozen water in a fractured specimen is evaporated from the surface before shadowing, three-dimensional details of deeper parts of the cytoplasm can be revealed. A variation of this method involves extracting soluble molecules and membranes with mild detergents before freezing, fracturing, evaporating frozen water, and rotary-shadowing (Fig. 6-4 E; see also Fig. 1-13).
A scanning electron microscope (SEM) can be used on thicker specimens, such as whole cells or tissues that have been fixed, dried, and coated with a thin metal film. Here, an electron beam scans a raster pattern over the surface of specimens, and secondary electrons emitted from the surface at each point are collected and used to reconstruct an image (Fig. 6-4 A). The resolution of conventional SEM is limited, but nonetheless valuable, for studying surface features of cells and their three-dimensional relationships in tissues. SEMs that use special high-energy (field emission) guns to produce the electron beam have greatly improved resolution, and these have been very useful for studying cellular substructures, such as nuclear pores (see Fig. 14-6B).
Choice of Organisms for Biological Research
Given the origin of life from a common ancestor (see Fig. 2-1), one can learn about basic cellular processes in any organism that has the molecules of interest. It is useful to select an organism that specializes in the process, such as skeletal muscle to study contractile proteins (see Chapter 39) or Chlamydomonas to study flagella (see Fig. 38-20). Some organisms are much more amenable to investigation because communities of scientists have invested years of hard work to develop genetic, molecular genetic, and biochemical methods for experimentation. These valuable experimental tools have attracted investigators to a growing number of “model” organisms (Table 6-2).
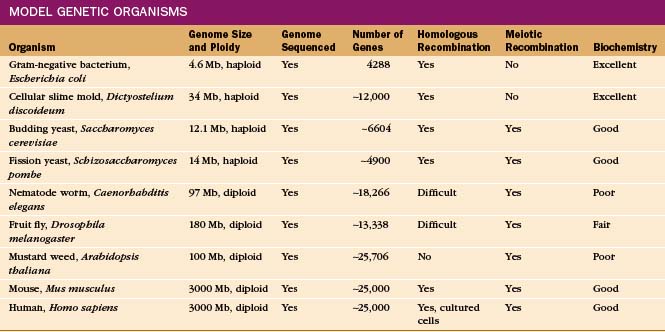
Model Organisms
Ideal model organisms have completely sequenced genomes and facile methods to manipulate the genes, including replacement of a gene with a modified gene, by the process of homologous recombination. Haploid organisms with one copy of each chromosome after mitotic division are particularly favorable for detecting the effects of changes in genes, called mutations (Box 6-2). It is useful for a haploid organism to have a diploid stage with two copies of each chromosome and a sexual phase, during which meiotic recombination occurs between the chromosomes from the two parents. (See Fig. 45-7 for details on recombination.) This allows one to construct strains with a variety of mutations and facilitates mapping mutations to a particular gene. In addition, diploids carrying a lethal mutation of a gene that is essential for life can be propagated, provided that the mutation is recessive.
BOX 6-2 Key Genetic Terms
Complementation. Providing gene function in trans (i.e., by another copy of a gene)
Diploid. A genome with two copies of each chromosome, one from each parent
Essential Gene. A gene whose function is required for viability
Genome. The entire genetic endowment of an or-ganism
Genotype. The genetic complement, including particular mutations
Haploid. A genome with single copies of each chro-mosome
Mutant. An organism that contains a mutation of interest
Pedigree. Family history of a genetic trait
Budding yeast and fission yeast meet all of these criteria, so they are widely used to study basic cellular functions. These free-living haploid organisms have a tractable diploid stage in their life cycles. Moving between haploid and diploid stages greatly simplifies the process of creating and analyzing recessive mutations. This is important because most loss-of-function mutations are recessive. Even before their genomes were sequenced, the availability of yeast for genetic, biochemical, and microscopic analysis revolutionized research in cell biology. However, yeast are solitary cells with specialized lifestyles.



