OBJECTIVES
After studying this chapter, you should be able to:
Name the key hormones secreted by Leydig cells and Sertoli cells of the testes and by Graafian follicles and corpora lutea of the ovaries.
Outline the role of chromosomes, hormones, and related factors in sex determination and development.
Summarize the hormonal changes that occur at puberty in males and females.
Outline the hormonal changes and their physiologic effects during perimenopause and menopause.
Describe the physiologic changes that occur in the female reproductive organs during the menstrual cycle.
Know the general structures of 17β-estradiol and progesterone, and describe their biosynthesis, transport, metabolism, and actions.
Describe the roles of the pituitary and the hypothalamus in the regulation of ovarian function, and the role of feedback loops in this process.
Describe the hormonal changes that accompany pregnancy and parturition.
Outline the processes involved in lactation.
INTRODUCTION
Modern genetics and experimental embryology make it clear that, in most species of mammals, the multiple differences between the male and the female depend primarily on a single chromosome (the Y chromosome) and a single pair of endocrine structures, namely the testes in the male and the ovaries in the female. The differentiation of the primitive gonads into testes or ovaries in utero is genetically determined in humans, but the formation of male genitalia depends on the presence of a functional, secreting testis; in the absence of testicular tissue, development is female. Evidence indicates that male sexual behavior and, in some species, the male pattern of gonadotropin secretion are due to the action of male hormones on the brain in early development. After birth, the gonads remain quiescent until adolescence, when they are activated by gonadotropins from the anterior pituitary. Hormones secreted by the gonads at this time cause the appearance of features typical of the adult male or female and the onset of the sexual cycle in the female. In human females, ovarian function regresses after a number of years and sexual cycles cease (the menopause). In males, gonadal function slowly declines with advancing age, but the ability to produce viable gametes persists.
In both sexes, the gonads have a dual function: the production of germ cells (gametogenesis) and the secretion of sex hormones. The androgens are steroid sex hormones that are masculinizing in their action; the estrogens are those that are feminizing. Both types of hormones are normally secreted in both sexes. The ovaries secrete large amounts of estrogens and small amounts of androgens, a pattern that is reversed in males. Androgens are secreted from the adrenal cortex in both sexes, and some of the androgens are converted to estrogens in fat and other extragonadal and extra-adrenal tissues. The ovaries also secrete progesterone, a steroid that has special functions in preparing the uterus for pregnancy.
Particularly during pregnancy, the ovaries secrete the polypeptide hormone relaxin, which loosens the ligaments of the pubic symphysis and softens the cervix, facilitating delivery of the fetus. In both sexes, the gonads secrete other polypeptides, including inhibin B, a polypeptide that inhibits follicle-stimulating hormone (FSH) secretion.
The secretory and gametogenic functions of the gonads are both dependent on the secretion of the anterior pituitary gonadotropins, FSH, and luteinizing hormone (LH). The sex hormones and inhibin B feed back to inhibit gonadotropin secretion. In males, gonadotropin secretion is noncyclic; but in postpubertal females an orderly, sequential secretion of gonadotropins is necessary for the occurrence of menstruation, pregnancy, and lactation.
SEX DIFFERENTIATION & DEVELOPMENT
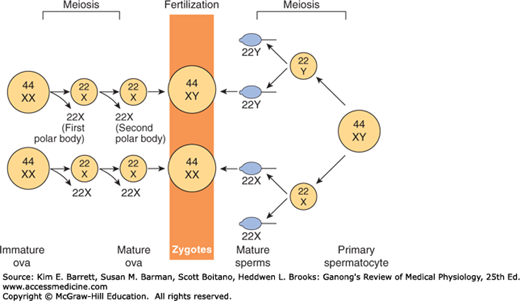
Sex is determined genetically by two chromosomes, called the sex chromosomes, to distinguish them from the somatic chromosomes (autosomes). In humans and many other mammals, the sex chromosomes are called X and Y. The Y chromosome is necessary and sufficient for the production of testes, and the testis-determining gene product is called SRY (for sex-determining region of the Y chromosome). SRY is a DNA-binding regulatory protein. It bends the DNA and acts as a transcription factor that initiates transcription of a cascade of genes necessary for testicular differentiation, including the gene for müllerian inhibiting substance (MIS; see below). The gene for SRY is located near the tip of the short arm of the human Y chromosome. Diploid male cells contain an X and a Y chromosome (XY pattern), whereas female cells contain two X chromosomes (XX pattern). As a consequence of meiosis during gametogenesis, each normal ovum contains a single X chromosome, but half of the normal sperm contain an X chromosome and half contain a Y chromosome (Figure 22–1). When a sperm containing a Y chromosome fertilizes an ovum, an XY pattern results and the zygote develops into a genetic male. When fertilization occurs with an X-containing sperm, an XX pattern and a genetic female results. Cell division and the chemical nature of chromosomes are discussed in Chapter 1.
FIGURE 22–1
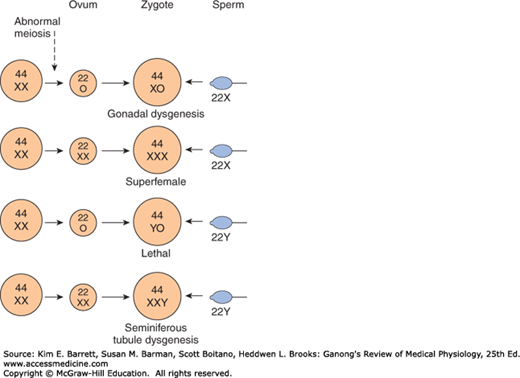
Basis of genetic sex determination. In the two-stage meiotic division in the female, only one cell survives as the mature ovum. In the male, the meiotic division results in the formation of four sperms, two containing the X and two the Y chromosome. Fertilization thus produces a male zygote with 22 pairs of autosomes plus an X and a Y or a female zygote with 22 pairs of autosomes and two X chromosomes. Note that for clarity, this figure and Figures 25–6 and 25–7 differ from the current international nomenclature for karyotypes, which lists the total number of chromosomes followed by the sex chromosome pattern. Thus, XO is 45, X; XY is 46, XY; XXY is 47, XXY, and so on.
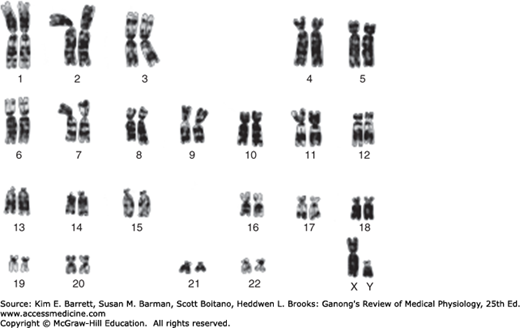
Human chromosomes can be studied in detail. Human cells are grown in tissue culture; treated with the drug colchicine, which arrests mitosis at the metaphase; exposed to a hypotonic solution that makes the chromosomes swell and disperse; and then “squashed” onto slides. Staining techniques make it possible to identify the individual chromosomes (Figure 22–2). There are 46 chromosomes: in males, 22 pairs of autosomes plus an X chromosome and a Y chromosome; in females, 22 pairs of autosomes plus two X chromosomes. The individual chromosomes are usually arranged in an arbitrary pattern (karyotype). The individual autosome pairs are identified by the numbers 1–22 on the basis of their morphologic characteristics.
Soon after cell division has started during embryonic development, one of the two X chromosomes of the somatic cells in normal females becomes functionally inactive. In abnormal individuals with more than two X chromosomes, only one remains active. The process that is normally responsible for inactivation is initiated in an X-inactivation center in the chromosome, probably via the transactivating factor CTCF (for CCCTC-binding factor), which is also induced during gene imprinting. However, the details of the inactivation process are still incompletely understood. The choice of which X chromosome remains active is random, so normally one X chromosome remains active in approximately half of the cells and the other X chromosome is active in the other half. The selection persists through subsequent divisions of these cells, and consequently some of the somatic cells in adult females contain an active X chromosome of paternal origin and some contain an active X chromosome of maternal origin.
In normal cells, the inactive X chromosome condenses and can be seen in various types of cells, usually near the nuclear membrane, as the Barr body, also called sex chromatin (Figure 22–3). Thus, there is a Barr body for each X chromosome in excess of one in the cell. The inactive X chromosome is also visible as a small “drumstick” of chromatin projecting from the nuclei of 1–15% of the polymorphonuclear leukocytes in females but not in males (Figure 22–3).
FIGURE 22–3
Left: Barr body (arrows) in the epidermal spinous cell layer. Right: Nuclear appendage (“drumstick”) identified by arrow in white blood cells. (Reproduced with permission from Grumbach MM, Barr ML: Cytologic tests of chromosomal sex in relation to sex anomalies in man. Recent Prog Horm Res 1958;14:255–324.)

On each side of the embryo, a primitive gonad arises from the genital ridge, a condensation of tissue near the adrenal gland. The gonad develops a cortex and a medulla. Until the sixth week of development, these structures are identical in both sexes. In genetic males, the medulla develops during the seventh and eighth weeks into a testis, and the cortex regresses. Leydig and Sertoli cells appear, and testosterone and MIS are secreted. In genetic females, the cortex develops into an ovary and the medulla regresses. The embryonic ovary does not secrete hormones. Hormonal treatment of the mother has no effect on gonadal (as opposed to ductal and genital) differentiation in humans, although it does in some experimental animals.
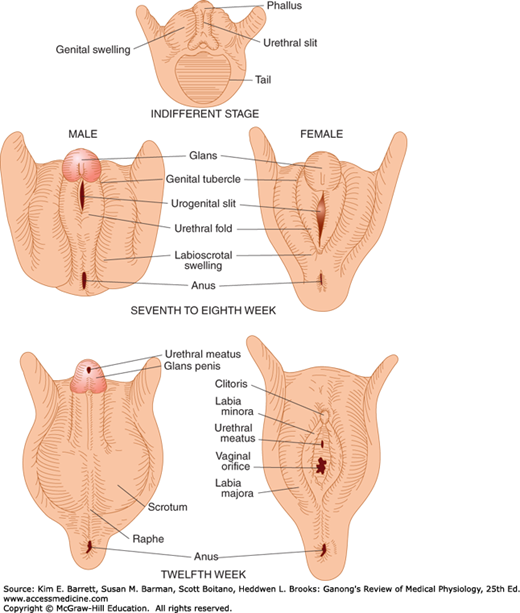
The embryology of the gonads is summarized in Figures 22–4 and 22–5. In the seventh week of gestation, the embryo has both male and female primordial genital ducts (Figure 22–4). In a normal female fetus, the müllerian duct system then develops into uterine tubes (oviducts) and a uterus. In the normal male fetus, the wolffian duct system on each side develops into the epididymis and vas deferens. The external genitalia are similarly bipotential until the eighth week (Figure 22–5). Thereafter, the urogenital slit disappears and male genitalia form, or, alternatively, it remains open and female genitalia form.

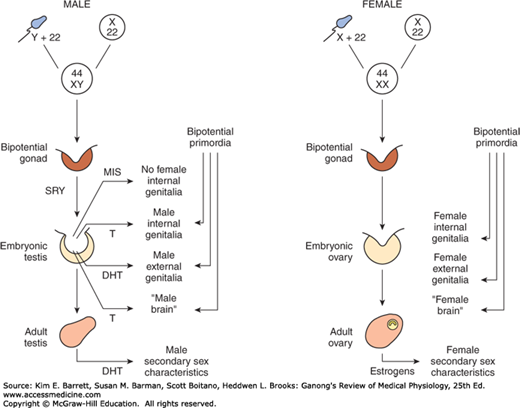
When the embryo has functional testes, male internal and external genitalia develop. The Leydig cells of the fetal testis secrete testosterone, and the Sertoli cells secrete MIS (also called müllerian regression factor, or MRF). MIS is a 536-amino-acid homodimer that is a member of the transforming growth factor β (TGF-β) super-family of growth factors, which includes inhibins and activins.
In their effects on the internal as opposed to the external genitalia, MIS and testosterone act unilaterally. MIS causes regression of the müllerian ducts by apoptosis on the side on which it is secreted, and testosterone fosters the development of the vas deferens and related structures from the wolffian ducts. The testosterone metabolite dihydrotestosterone induces the formation of male external genitalia and male secondary sex characteristics (Figure 22–6).
MIS continues to be secreted by the Sertoli cells, and it reaches mean values of 48 ng/mL in plasma in 1- to 2-year-old boys. Thereafter, it declines to low levels by the time of puberty and persists at low but detectable levels throughout life. In girls, MIS is produced by granulosa cells in small follicles in the ovaries, but plasma levels are very low or undetectable until puberty. Thereafter, plasma MIS is about the same as in adult men, that is, about 2 ng/mL. The functions of MIS after early embryonic life are unsettled, but it is probably involved in germ cell maturation in both sexes and in control of testicular descent in boys.
At least in some species, the development of the brain as well as the external genitalia is affected by androgens early in life. In rats, a brief exposure to androgens during the first few days of life causes the male pattern of sexual behavior and the male pattern of hypothalamic control of gonadotropin secretion to develop after puberty. In the absence of androgens, female patterns develop (see Chapter 17). In monkeys, similar effects on sexual behavior are produced by exposure to androgens in utero, but the pattern of gonadotropin secretion remains cyclical. Early exposure of female human fetuses to androgens also appears to cause subtle but significant masculinizing effects on behavior. However, women with adrenogenital syndrome due to congenital adrenocortical enzyme deficiency (see Chapter 20) develop normal menstrual cycles when treated with cortisol. Thus, the human, like the monkey, appears to retain the cyclical pattern of gonadotropin secretion despite exposure to androgens in utero.
From the preceding discussion, it might be expected that abnormalities of sexual development could be caused by genetic or hormonal abnormalities as well as by other nonspecific teratogenic influences, and this is indeed the case. The major classes of abnormalities are listed in Table 22–1.
| Chromosomal disorders |
| Gonadal dysgenesis (XO and variants) |
| “Superfemales” (XXX) |
| Seminiferous tubule dysgenesis (XXY and variants) |
| True hermaphroditism |
| Developmental disorders |
| Female pseudohermaphroditism |
| Congenital virilizing adrenal hyperplasia of fetus |
| Maternal androgen excess |
| Virilizing ovarian tumor |
| Iatrogenic: Treatment with androgens or certain synthetic progestational drugs |
| Male pseudohermaphroditism |
| Androgen resistance |
| Defective testicular development |
| Congenital 17α-hydroxylase deficiency |
| Congenital adrenal hyperplasia due to blockade of pregnenolone formation |
| Various nonhormonal anomalies |
Nondisjunction of sex chromosomes during the first division in meiosis results in distinct defects (Clinical Box 22–1; Figure 22–7). Meiosis is a two-stage process, and although nondisjunction usually occurs during the first meiotic division, it can occur in the second, producing more complex chromosomal abnormalities. In addition, nondisjunction or simple loss of a sex chromosome can occur during the early mitotic divisions after fertilization. The consequence of faulty mitoses in the early zygote is mosaicism, in which two or more populations of cells have different chromosome complements. True hermaphroditism, the condition in which the individual has both ovaries and testes, is probably due to XX/XY mosaicism and related mosaic patterns, although other genetic aberrations are possible.
Chromosomal abnormalities also include transposition of parts of chromosomes to other chromosomes. Rarely, genetic males are found to have the XX karyotype because the short arm of their father’s Y chromosome was transposed to their father’s X chromosome during meiosis and they received that X chromosome along with their mother’s. Similarly, deletion of the small portion of the Y chromosome containing SRY produces females with the XY karyotype.
Development of the male external genitalia occurs normally in genetic males in response to androgen secreted by the embryonic testes, but male genital development may also occur in genetic females exposed to androgens from some other source during the 8th to the 13th weeks of gestation. The syndrome that results is female pseudohermaphroditism. A pseudohermaphrodite is an individual with the genetic constitution and gonads of one sex and the genitalia of the other. After the 13th week, the genitalia are fully formed, but exposure to androgens can cause hypertrophy of the clitoris. Female pseudohermaphroditism may be due to congenital virilizing adrenal hyperplasia (see Chapter 20), or it may be caused by androgens administered to the mother. Conversely, one cause of the development of female external genitalia in genetic males (male pseudohermaphroditism) is defective testicular development. Because the testes also secrete MIS, genetic males with defective testes have female internal genitalia.
Another cause of male pseudohermaphroditism is androgen resistance, in which, as a result of various congenital abnormalities, male hormones cannot exert their full effects on the tissues. One form of androgen resistance is a 5α-reductase deficiency, in which the enzyme responsible for the formation of dihydrotestosterone, the active form of testosterone, is decreased (Figure 22–8). The consequences of this deficiency are discussed in Chapter 23. Other forms of androgen resistance are due to various mutations in the androgen receptor gene, and the resulting defects in receptor function range from minor to severe. Mild defects cause infertility with or without gynecomastia. When the loss of receptor function is complete, the testicular feminizing syndrome, now known as complete androgen resistance syndrome, results. In this condition, MIS is present and testosterone is secreted at normal or even elevated rates. The external genitalia are female, but the vagina ends blindly because there are no female internal genitalia. At puberty, enlarged breasts develop in persons with this syndrome. The syndrome usually remains undiagnosed until women seek medical advice because of lack of menstruation.
CLINICAL BOX 22–1 Chromosomal Abnormalities
An established defect in gametogenesis is nondisjunction, a phenomenon in which a pair of chromosomes fail to separate, so that both go to one of the daughter cells during meiosis. Four of the abnormal zygotes that can form as a result of nondisjunction of one of the X chromosomes during oogenesis are shown in Figure 25–7. In individuals with the XO chromosomal pattern, the gonads are rudimentary or absent, so that female external genitalia develop, stature is short, other congenital abnormalities are often present, and no sexual maturation occurs at puberty. This syndrome is called gonadal dysgenesis or, alternatively, ovarian agenesis or Turner syndrome. Individuals with the XXY pattern, the most common sex chromosome disorder, have the genitalia of a normal male. Testosterone secretion at puberty is often great enough for the development of male characteristics; however, the seminiferous tubules are abnormal, and the incidence of mental retardation is higher than normal. This syndrome is known as seminiferous tubule dysgenesis or Klinefelter syndrome. The XXX (“superfemale”) pattern is second in frequency only to the XXY pattern and may be even more common in the general population, since it does not seem to be associated with any characteristic abnormalities. The YO combination is probably lethal.
Nondisjunction of chromosome 21 produces trisomy 21, the chromosomal abnormality associated with Down syndrome. The additional chromosome 21 is normal, so Down syndrome is a pure case of gene excess causing abnormalities.
Many other chromosomal abnormalities occur as well as numerous diseases due to defects in single genes. These conditions are generally diagnosed in utero by analysis of fetal cells in a sample of amniotic fluid collected by inserting a needle through the abdominal wall (amniocentesis) or, earlier in pregnancy, by examining fetal cells obtained by a needle biopsy of chorionic villi (chorionic villus sampling).
THERAPEUTIC HIGHLIGHTSMany of the syndromes mentioned have effects on multiple organ systems, and patients must be carefully monitored with a multidisciplinary approach to avert the consequences of cardiovascular defects, infections secondary to urinary tract and renal malformations, and the psychological impact of reproductive implications. Girls with Turner syndrome and evidence of gonadal failure are also treated with low-dose estrogen to evoke puberty, followed by gradual replacement of mature estrogen levels to permit feminization. Conversely, patients with Klinefelter syndrome are often supplemented with androgens to improve virilization and libido.
It is worth noting that genetic males with congenital blockage of the formation of pregnenolone are pseudohermaphrodites because testicular as well as adrenal androgens are normally formed from pregnenolone. Male pseudohermaphroditism also occurs when there is a congenital deficiency of 17α-hydroxylase (see Chapter 20).
As noted above, a burst of testosterone secretion occurs in male fetuses before birth (see Chapter 23). In the neonatal period there is another burst, with unknown function, but thereafter the Leydig cells become quiescent. There follows in all mammals a period in which the gonads of both sexes are quiescent until they are activated by gonadotropins from the pituitary to bring about the final maturation of the reproductive system. This period of final maturation is known as adolescence. It is often also called puberty, although puberty, strictly defined, is the period when the endocrine and gametogenic functions of the gonads have first developed to the point where reproduction is possible. In girls, the first event is thelarche, the development of breasts, followed by pubarche, the development of axillary and pubic hair, and then by menarche, the first menstrual period. Initial menstrual periods are generally anovulatory, and regular ovulation appears about a year later. In contrast to the situation in adulthood, removal of the gonads during the period from soon after birth to puberty causes only a small increase in gonadotropin secretion, so gonadotropin secretion is not being held in check by the gonadal hormones. In children between the ages of 7 and 10, a slow increase in estrogen and androgen secretion precedes the more rapid rise in the early teens (Figure 22–9).
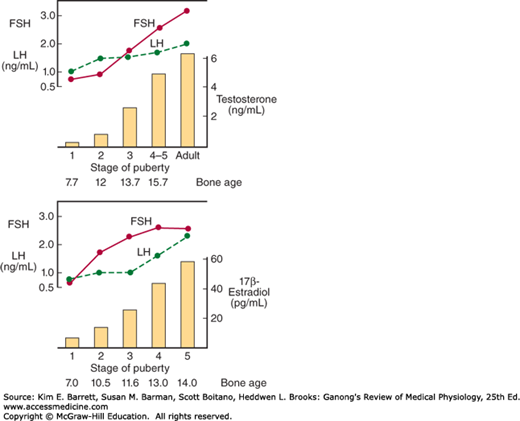
FIGURE 22–9
Changes in plasma hormone concentrations during puberty in boys (top) and girls (bottom). Stage 1 of puberty is preadolescence in both sexes. In boys, stage 2 is characterized by beginning enlargement of the testes, stage 3 by penile enlargement, stage 4 by growth of the glans penis, and stage 5 by adult genitalia. In girls, stage 2 is characterized by breast buds, stage 3 by elevation and enlargement of the breasts, stage 4 by projection of the areolas, and stage 5 by adult breasts. FSH, follicle-stimulating hormone; LH, luteinizing hormone. (Modified and reproduced with permission from Berenberg SR [ed]: Puberty: Biologic and Psychosocial Components. HE Stenfoert Kroese BV; 1975.)
The age at the time of puberty is variable. In Europe and the United States, it has been declining at the rate of 1–3 months per decade for more than 175 years. In the United States in recent years, puberty generally occurs between the ages of 8 and 13 in girls and 9 and 14 in boys.
Another event that occurs in humans at the time of puberty is an increase in the secretion of adrenal androgens (see Figure 20–12). The onset of this increase is called adrenarche. It occurs at age 8–10 years in girls and age 10–12 years in boys. Dehydroepiandrosterone (DHEA) values peak at about age 25 in women and slightly later than that in men. They then decline slowly to low values in old age. The rise appears to be due to an increase in the activity of 17α-hydroxylase.
The gonads of children can be stimulated by gonadotropins; their pituitaries contain gonadotropins and their hypothalami contain gonadotropin-releasing hormone (GnRH) (see Chapter 17). However, their gonadotropins are not secreted. In immature monkeys, normal menstrual cycles can be brought on by pulsatile injection of GnRH, and they persist as long as the pulsatile injection is continued. Thus, it seems clear that pulsatile secretion of GnRH brings on puberty. During the period from birth to puberty, a neural mechanism is operating to prevent the normal pulsatile release of GnRH. The nature of the mechanism inhibiting the GnRH pulse generator is unknown. However, one or more genes produce products that stimulate secretion of GnRH, and inhibition of these genes before puberty is an interesting possibility (Clinical Box 22–2).
The major causes of precocious sexual development in humans are listed in Table 22–2. Early development of secondary sexual characteristics without gametogenesis is caused by abnormal exposure of immature males to androgen or females to estrogen. This syndrome should be called precocious pseudopuberty to distinguish it from true precocious puberty due to an early but otherwise normal pubertal pattern of gonadotropin secretion from the pituitary.
| True precocious puberty |
| Constitutional |
| Cerebral: Disorders involving posterior hypothalamus |
| Tumors |
| Infections |
| Developmental abnormalities |
| Gonadotropin-independent precocity |
| Precocious pseudopuberty (no spermatogenesis or ovarian development) |
| Adrenal |
| Congenital virilizing adrenal hyperplasia |
| Androgen-secreting tumors (in males) |
| Estrogen-secreting tumors (in females) |
| Gonadal |
| Leydig cell tumors of testis |
| Granulosa cell tumors of ovary |
| Miscellaneous |
Constitutional precocious puberty, that is, precocious puberty in which no cause can be determined, is more common in girls than in boys. In both sexes, tumors or infections involving the hypothalamus cause precocious puberty. Indeed, in one large series of cases, precocious puberty was the most common endocrine symptom of hypothalamic disease. In experimental animals, precocious puberty can be produced by hypothalamic lesions. Apparently the lesions interrupt a pathway that normally holds pulsatile GnRH secretion in check. Pineal tumors are sometimes associated with precocious puberty, but evidence indicates that these tumors are associated with precocity only when there is secondary damage to the hypothalamus.
CLINICAL BOX 22–2 Leptin
It has been argued for some time that a critical body weight must normally be reached for puberty to occur. Thus, for example, young women who engage in strenuous athletics lose weight and stop menstruating, as do girls with anorexia nervosa. If these girls start to eat and gain weight, they menstruate again, that is, they “go back through puberty.” It now appears that leptin, the satiety-producing hormone secreted by fat cells, may be the link between body weight and puberty. Obese ob/ob mice that cannot make leptin are infertile, and their fertility is restored by injections of leptin. Leptin treatment also induces precocious puberty in immature female mice. However, the way that leptin fits into the overall control of puberty remains to be determined.
Precocious gametogenesis and steroidogenesis can occur without the pubertal pattern of gonadotropin secretion (gonadotropin-independent precocity). At least in some cases of this condition, the sensitivity of LH receptors to gonadotropins is increased because of an activating mutation in the G-protein that couples the receptors to adenylyl cyclase.
CLINICAL BOX 22–3 Hyperprolactinemia
Up to 70% of the patients with chromophobe adenomas of the anterior pituitary have elevated plasma prolactin levels. In some instances, the elevation may be due to damage to the pituitary stalk, but in most cases, the tumor cells are actually secreting the hormone. The hyperprolactinemia may cause galactorrhea, but in many individuals no demonstrable endocrine abnormalities are present. Conversely, most women with galactorrhea have normal prolactin levels; definite elevations are found in less than one-third of patients with this condition.
Another interesting observation is that 15–20% of women with secondary amenorrhea have elevated prolactin levels, and when prolactin secretion is reduced, normal menstrual cycles and fertility return. Prolactin may produce amenorrhea by blocking the action of gonadotropins on the ovaries. The hypogonadism produced by prolactinomas is associated with osteoporosis due to estrogen deficiency.
As noted previously, hyperprolactinemia in men is associated with erectile dysfunction and hypogonadism that disappear when prolactin secretion is reduced.
THERAPEUTIC HIGHLIGHTSPrescription drug use is a common cause of hyperprolactinemia. Prolactin secretion in the pituitary is suppressed by the brain chemical dopamine. Use of drugs that block the effects of dopamine can cause the pituitary to secrete prolactin. Examples of some prescription drugs that can cause hyperprolactinemia include haloperidol and phenothiazines; most antipsychotic medications; and cisapride, which is used to treat nausea and gastroesophageal reflux in cancer patients. If possible, the drug suspected as causing hyperprolactinemia should be withdrawn, or the dose titrated. Regardless of the etiology, treatment should endeavor to restore normal prolactin levels to avoid suppressive effects on the ovaries and preserve bone density. Dopamine agonists also provide benefit in many cases, including prolactinoma, and can be used in patients for whom an inciting pharmaceutical agent cannot be withdrawn.
The normal variation in the age at which adolescent changes occur is so wide that puberty cannot be considered to be pathologically delayed until the menarche has failed to occur by the age of 17 or testicular development by the age of 20. Failure of maturation due to panhypopituitarism is associated with dwarfing and evidence of other endocrine abnormalities. Patients with the XO chromosomal pattern and gonadal dysgenesis are also dwarfed. In some individuals, puberty is delayed even though the gonads are present and other endocrine functions are normal. In males, this clinical picture is called eunuchoidism. In females, it is called primary amenorrhea (Clinical Box 22–3).
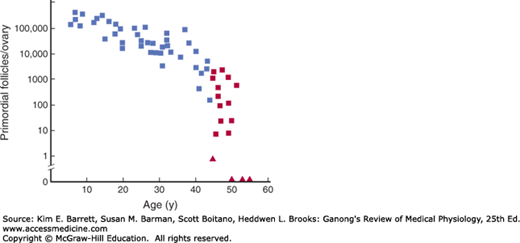
The human ovaries become unresponsive to gonadotropins with advancing age, and their function declines, so that sexual cycles disappear (menopause). This unresponsiveness is associated with and probably caused by a decline in the number of primordial follicles, which becomes precipitous at the time of menopause (Figure 22–10). The ovaries no longer secrete progesterone and 17β-estradiol in appreciable quantities, and estrogen is formed only in small amounts by aromatization of androstenedione in peripheral tissues (see Chapter 20). The uterus and the vagina gradually become atrophic. As the negative feedback effect of estrogens and progesterone is reduced, secretion of FSH is increased, and plasma FSH increases to high levels, LH levels are moderately high. Old female mice and rats have long periods of diestrus and increased levels of gonadotropin secretion. In women, a period called perimenopause precedes menopause and can last up to 10 years. During perimenopause FSH levels will increase before an increase in LH is observed due to a decrease in estrogen, progesterone, and inhibins and the menses become irregular. This usually occurs between the ages of 45 and 55. The average age at onset of the menopause is 52 years.
FIGURE 22–10
Number of primordial follicles per ovary in women at various ages. Blue squares, premenopausal women (regular menses); red squares, perimenopausal women (irregular menses for at least 1 year); red triangles, postmenopausal women (no menses for at least 1 year). Note that the vertical scale is a log scale and that the values are from one rather than two ovaries. (Reproduced with permission from Richardson SJ, Senikas V, Nelson JF: Follicular depletion during the menopausal transition: Evidence for accelerated loss and ultimate exhaustion. J Clin Endocrinol Metab 1987; Dec; 65(6):1231–1237.)
The loss of ovarian function causes many symptoms such as sensations of warmth spreading from the trunk to the face (hot flushes; also called hot flashes) and night sweats. In addition, the onset of menopause increases the risk of many diseases such as osteoporosis, ischemic heart disease, and renal disease.
Hot flushes are said to occur in 75% of menopausal women and may continue intermittently for as long as 40 years. They also occur when early menopause is produced by bilateral ovariectomy, and they are prevented by estrogen treatment. In addition, they occur after castration in men. Their cause is unknown. However, they coincide with surges of LH secretion. LH is secreted in episodic bursts at intervals of 30–60 min or more (circhoral secretion), and in the absence of gonadal hormones these bursts are large. Each hot flush begins with the start of a burst. However, LH itself is not responsible for the symptoms, because they can continue after removal of the pituitary. Instead, it appears that some estrogen-sensitive event in the hypothalamus initiates both the release of LH and the episode of flushing.
Although the function of the testes tends to decline slowly with advancing age, the evidence is unclear whether there is a “male menopause” (andropause) similar to that occurring in women.
THE FEMALE REPRODUCTIVE SYSTEM
The reproductive system of women (Figure 22–11), unlike that of men, shows regular cyclic changes that teleologically may be regarded as periodic preparations for fertilization and pregnancy. In humans and other primates, the cycle is a menstrual cycle, and its most conspicuous feature is the periodic vaginal bleeding that occurs with the shedding of the uterine mucosa (menstruation).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


