Renal Cell Carcinoma
KEY CONCEPTS
![]() Renal cell carcinoma (RCC) predominantly occurs later in life, with 70% of all cases diagnosed between the ages of 55 and 84 years.
Renal cell carcinoma (RCC) predominantly occurs later in life, with 70% of all cases diagnosed between the ages of 55 and 84 years.
![]() The most established risk factors for RCC are smoking, obesity, and hypertension.
The most established risk factors for RCC are smoking, obesity, and hypertension.
![]() Inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene is the hallmark of the most common type of RCC, the clear cell subtype.
Inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene is the hallmark of the most common type of RCC, the clear cell subtype.
![]() More than 50% of RCC cases are diagnosed by incidental findings on routine imaging for unrelated reasons.
More than 50% of RCC cases are diagnosed by incidental findings on routine imaging for unrelated reasons.
![]() The Memorial Sloan-Kettering Cancer Center Prognostic Factors Model for Survival classifies patients into low-, intermediate-, and high-risk groups based on five clinical factors and can predict survival in untreated patients and those treated with immunotherapy and targeted agents.
The Memorial Sloan-Kettering Cancer Center Prognostic Factors Model for Survival classifies patients into low-, intermediate-, and high-risk groups based on five clinical factors and can predict survival in untreated patients and those treated with immunotherapy and targeted agents.
![]() Surgical removal of the primary tumor, either by total or partial nephrectomy, is the preferred initial treatment for all stages of RCC.
Surgical removal of the primary tumor, either by total or partial nephrectomy, is the preferred initial treatment for all stages of RCC.
![]() Immunotherapy used to be considered first-line therapy for metastatic RCC but has largely been replaced by targeted agents because of their improved efficacy and tolerability.
Immunotherapy used to be considered first-line therapy for metastatic RCC but has largely been replaced by targeted agents because of their improved efficacy and tolerability.
![]() Sunitinib and pazopanib are oral small molecule inhibitors of vascular endothelial growth factor (VEGF) and platelet-derived growth factor and are each treatment options as first-line therapy for metastatic RCC.
Sunitinib and pazopanib are oral small molecule inhibitors of vascular endothelial growth factor (VEGF) and platelet-derived growth factor and are each treatment options as first-line therapy for metastatic RCC.
![]() The VEGFR–tyrosine kinase inhibitor axitinib and the mammalian target of rapamycin (mTOR) inhibitor everolimus are both second-line therapy options for metastatic RCC patients who progress on a targeted therapy or cytokine-based therapy first-line regimen.
The VEGFR–tyrosine kinase inhibitor axitinib and the mammalian target of rapamycin (mTOR) inhibitor everolimus are both second-line therapy options for metastatic RCC patients who progress on a targeted therapy or cytokine-based therapy first-line regimen.
![]() Temsirolimus is an IV administered mTOR inhibitor indicated for first-line therapy in patients with high-risk metastatic RCC.
Temsirolimus is an IV administered mTOR inhibitor indicated for first-line therapy in patients with high-risk metastatic RCC.
INTRODUCTION
Renal cell carcinoma (RCC) is a less common malignancy that, until recently, had few treatment options that were poorly tolerated and resulted in few positive outcomes for patients. However, treatment for the disease has been revolutionized by an increased understanding of the pathophysiology of RCC. Clear cell is the predominant subtype of RCC and is the result of inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene on chromosome 3p25, which leads to increased production of growth factors such as vascular endothelial growth factor (VEGF), transforming growth factor (TGF), platelet-derived growth factor (PDGF), and others responsible for angiogenesis and cell growth.1 Before 2005, the primary therapy option for patients with advanced RCC after nephrectomy was immunotherapy with few responses and high toxicity. However, seven new drugs have been approved as first- or second-line therapy for RCC: sorafenib, sunitinib, temsirolimus, bevacizumab (in combination with interferon-α [IFN-α]), everolimus, pazopanib, and axitinib.2–7 Each drug is an example of targeted therapy against growth factors important in the pathophysiology of RCC and has yielded much needed progress in a disease with few therapeutic options. RCC serves as an example of rational development of targeted agents based on knowledge of tumor biology for the treatment of other malignancies.
EPIDEMIOLOGY
About 65,000 new cases of kidney and renal pelvis cancer are diagnosed each year in the United States, with two-thirds of these cases occurring in men. More than 13,000 people in the United States will die of kidney cancer each year.8 RCC is the fifth most commonly occurring cancer in men, and the number of new cases diagnosed each year is similar to non-Hodgkin lymphoma and melanoma. In women, kidney cancer is the sixth most common cancer, occurring at a rate similar to the rates for ovarian and pancreatic cancers. The incidence of RCC has increased over the past three decades. The rate has increased more rapidly in blacks than whites and in women than men. In the United States, between 2002 and 2006, the age-adjusted incidence rate in black men was 21.3, white men 19.2, black women 10.3, and white women 9.9 per 100,000 person years.9 This increase may be related to improved imaging techniques and greater use of these imaging modalities, although the higher prevalence of some risk factors may also explain the increased incidence.
Kidney cancer is most commonly diagnosed between the ages of 40 and 70 years, with a peak in the sixth and seventh decades of life. Nearly 70% of all cases of kidney cancer are diagnosed in people between the ages of 55 and 84 years, with less than 3% of all cases diagnosed in patients younger than 34 years.9 ![]()
One of the primary factors influencing overall survival (OS) is the extent of disease spread. When the tumor is confined to the kidney at the time of diagnosis, surgical resection can result in a 5-year OS rate of about 85%. That figure falls to 64% when localized spread has occurred beyond the kidney and to less than 23% when metastatic disease is present.10 The 5-year relative survival rate of kidney cancer in general improved from 40% in the early 1960s to nearly 70% in 2005.9 Survival is expected to improve with the growing availability of numerous targeted agents.
ETIOLOGY
The incidence rates of RCC vary more than 10-fold worldwide, with the highest incidence rates in Western and European countries and the lowest in Asia and Africa, which suggests that lifestyle and possibly the environment are important factors. The most established risk factors associated with RCC are smoking, obesity, and hypertension.
![]() Smoking remains the most consistently established risk factor and is estimated to be responsible for 20% to 30% and 10% to 20% of RCC cases diagnosed in men and women, respectively.11,12 Smoking is associated with a relative risk of 1.54 for men and 1.22 for women with a strong dose-dependent relationship. Heavy smoking, defined as 21 or more cigarettes per day, was associated with an increased relative risk of 2.03 and 1.58 for men and women, respectively.13 Smoking cessation has been demonstrated to reduce the risk of RCC with a 15% to 30% decrease after 10 to 15 years after cessation and a 50% decrease for those who quit for 30 years or more.13,14
Smoking remains the most consistently established risk factor and is estimated to be responsible for 20% to 30% and 10% to 20% of RCC cases diagnosed in men and women, respectively.11,12 Smoking is associated with a relative risk of 1.54 for men and 1.22 for women with a strong dose-dependent relationship. Heavy smoking, defined as 21 or more cigarettes per day, was associated with an increased relative risk of 2.03 and 1.58 for men and women, respectively.13 Smoking cessation has been demonstrated to reduce the risk of RCC with a 15% to 30% decrease after 10 to 15 years after cessation and a 50% decrease for those who quit for 30 years or more.13,14
Obesity is also linked to RCC development in observational studies and appears to be equally associated in men and women. Compared with a body mass index (BMI) of 20.75 kg/m2 or less, men with a BMI between 22.86 and 27.75 kg/m2 had a relative risk of 1.3 to 1.7, and those with a BMI of 27.76 kg/m2 or greater had a relative risk of 1.9.15 A separate analysis reported a relative risk of 2.5 for those with a BMI of 30 kg/m2 or greater.16 It is estimated that 30% to 40% of RCC cases may be attributed to obesity, which suggests that increasing rates of obesity in the United States may be partially responsible for the increased incidence seen over the past 3 decades.12 Numerous mechanisms explaining the link between obesity and the development of RCC have been proposed. Obesity has been linked to increased lipid peroxidation, which can result in carcinogenesis of the proximal renal tubules. Byproducts of the lipid peroxidation pathway have also been shown to result in DNA adducts in the kidney, which can result in oncogene and tumor suppressor gene mutations and eventually malignancy.17 Adipose tissue, when stimulated, can release numerous substances to regulate energy balance and lipid metabolism. Insulin resistance and compensatory elevated insulin levels result in increased levels of insulin-like growth factor-1 (IGF-1), which regulates cell proliferation and can inhibit cellular apoptosis.16 Finally, the kidneys of obese men and women are more susceptible to carcinogenesis because of higher glomerular filtration rates, renal perfusion, and atrophic scarring of the kidneys.12
The risk of RCC is associated with increased duration and severity of elevated blood pressure. Patients with a diastolic blood pressure (DBP) greater than 90 mm Hg had a relative risk of 2.1 compared with DBP less than 70 mm Hg. A systolic blood pressure (SBP) greater than 150 mm Hg was associated with a relative risk of 1.6 compared with SBP less than 120 mm Hg.15 The exact mechanism explaining the causality of hypertension to RCC is unknown, but it is believed to be related to hypertension-induced renal injury and lipid peroxidation.12,17 Medications commonly used to treat hypertension do not appear to be associated with RCC.
The analgesic phenacetin has a historical link to RCC. The drug was introduced in 1887 and used until the 1970s, when increased concerns for carcinogenesis resulted in replacement with safer analgesics, such as the major metabolite of phenacetin, acetaminophen. Despite its association with the known carcinogen, acetaminophen has not been associated with an increased risk of RCC.12
A well-defined genetic link has also been described. Although most RCC cases are not associated with hereditary factors and are considered “sporadic,” 2% to 3% of cases are secondary to inherited syndromes.18,19 Kidney tumors arising from a genetic etiology are most commonly the result of an autosomal dominant transmission of the diseased gene from a carrier to the offspring. Initially, one carrier parent has one healthy chromosome and one chromosome with the diseased gene. When this carrier has offspring with a healthy individual with two healthy chromosomes, the offspring each have a 50% chance of also being a carrier. These carriers with one healthy chromosome and one chromosome with the diseased gene are then more sensitive to developing RCC after being exposed to additional somatic mutations that can affect the remaining healthy chromosome.
SUBTYPES AND PATHOPHYSIOLOGY
About 85% of kidney cancers affect the parenchyma of the kidney and are classified as RCC. The renal pelvis is less commonly affected, making up 12% of the cases of kidney cancer diagnosed each year, with other rare malignancies affecting other parts of the kidney and making up the remaining 3%. The subtypes of RCC include clear cell, papillary (also known as chromophilic), chromophobic, and oncocytic. Each subtype has a unique genetic pathophysiology that results in a different clinical course and response to therapy.18
Clear Cell Renal Cell Carcinoma: The Role of the von Hippel-Lindau Gene
Clear cell is the predominant subtype responsible for most cases of RCC. These tumors typically affect the proximal tubule of the kidney and are more likely to metastasize than other subtypes. The initial finding of an association between clear cell histology and losses in the short arm of chromosome 3 eventually led to the gene responsible for this subtype being mapped to the location 3p24-25 and termed the VHL gene in 1993.20–22 Inactivation of this tumor suppressor gene is now recognized as the hallmark of clear cell RCC. The Knudson and Strong two-hit model explains the development of clear cell RCC after inactivation of both copies of the tumor suppressor gene VHL.22 In patients with sporadic disease, the two copies of VHL present in a healthy kidney can be inactivated via loss of chromosome 3p, gene silencing via hypermethylation, missense mutations, and premature truncation or nonsense mutations. Additional mutations can result in a single, unilateral tumor. In patients with hereditary disease, one copy of VHL has already been deleted via a germline mutation. Fewer events are then needed to delete the remaining gene copy, which explains why patients with hereditary disease are more likely to present with multicentric, bilateral tumors.18,19
The VHL gene produces the VHL protein (pVHL), which is expressed ubiquitously throughout the body and is part of the complex that selects substances for ubiquitination and subsequent destruction by the proteasome.23 Because of this role, VHL regulates cellular response to oxygen. In a normal oxygen environment, the target for proteasome destruction is hypoxia-inducible factor (HIF)-1α. Hydroxylated HIF-1α binds to pVHL and is destroyed by the proteasome (Fig. 115-1). When the cellular environment is hypoxic, HIF-1α is not hydroxylated and does not bind to pVHL. The unbound HIF-1α can then initiate transcription of hypoxia-inducible genes in the cell nucleus, which enables the cell to adapt and survive a hypoxic insult23,24 (Fig. 115-2). ![]() In the case of clear cell RCC, when VHL is mutated or silenced, pVHL is unable to bind and target HIF-1α for degradation regardless of the oxygen presence in the environment. HIF-1α therefore becomes abundant and activates transcription of hypoxia-inducible genes, including VEGF, PDGF, TGF, glucose transporters, erythropoietin, and other factors that promote angiogenesis and tumor development23,24 (Fig. 115-3). Numerous therapies for RCC target these hypoxia-inducible genes that are activated by HIF and are discussed later in the chapter.
In the case of clear cell RCC, when VHL is mutated or silenced, pVHL is unable to bind and target HIF-1α for degradation regardless of the oxygen presence in the environment. HIF-1α therefore becomes abundant and activates transcription of hypoxia-inducible genes, including VEGF, PDGF, TGF, glucose transporters, erythropoietin, and other factors that promote angiogenesis and tumor development23,24 (Fig. 115-3). Numerous therapies for RCC target these hypoxia-inducible genes that are activated by HIF and are discussed later in the chapter.
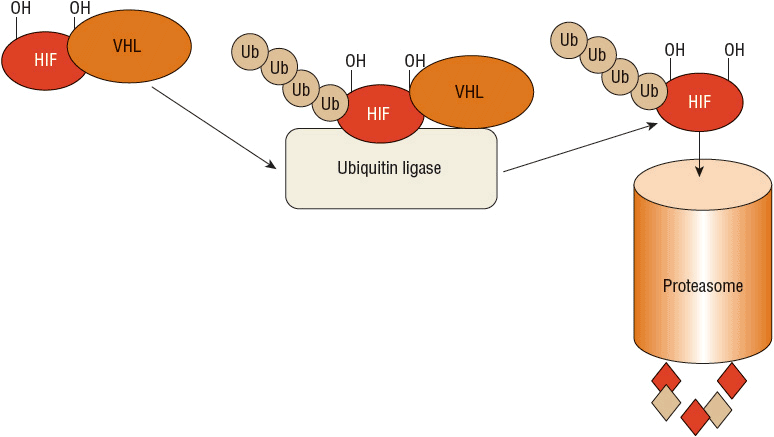
FIGURE 115-1 The role of von Hippel-Lindau (VHL) protein and hypoxia-inducible factor (HIF): normal oxygen, normal VHL. In a normal oxygen environment, HIF is hydroxylated. This enables binding of the VHL protein and subsequent attachment of a polyubiquitin chain, a process called ubiquitination. This allows the ubiquitin-tagged HIF to be recognized for destruction by the proteasome. The proteasome acts as a garbage disposal for compounds labeled by the ubiquitination process.
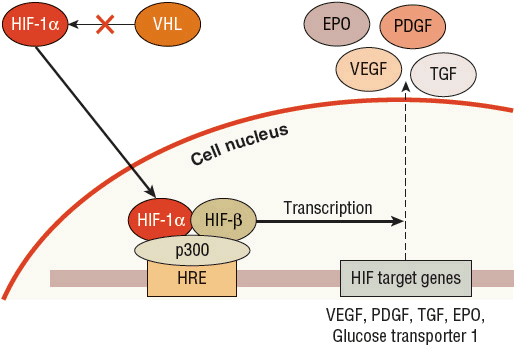
FIGURE 115-2 The role of von Hippel-Lindau (VHL) and hypoxia-inducible factor (HIF)-1α: low oxygen, normal VHL. In a low oxygen environment, the cell wants to increase production of substances to promote a switch to anaerobic metabolism, including enzymes involved in glycolysis and glycerol metabolism. In this situation, HIF-1α is not hydroxylated and cannot bind to VHL. HIF-1α is then able to translocate into the nucleus of the cell. In the nucleus, HIF-1α combines with the HIF-β subunit and the p300 transcriptional cofactor on the hypoxia response element (HRE) that promotes the transcription of HIF-1α target genes. More than 100 genes can be activated by this complex and include vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), transforming growth factor (TGF), erythropoietin (EPO), and glucose transporter 1.
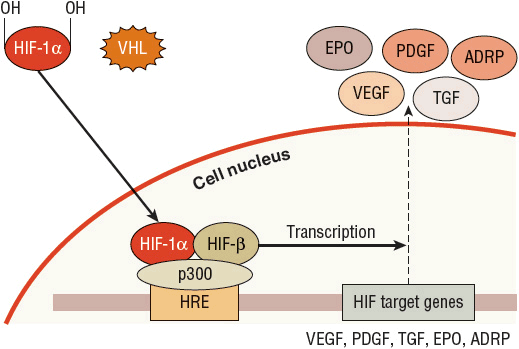
FIGURE 115-3 The role of von Hippel-Lindau (VHL) and hypoxia-inducible factor (HIF)-1α: normal oxygen, mutated VHL. When VHL is mutated, it is not able to bind to the hydroxylated HIF-1α regardless of the presence of oxygen in the environment. Because HIF-1α is not bound to VHL, it is not destroyed by the proteasome and is thus free to translocate into the nucleus, combine with the HIF-β subunit and the p300 transcriptional cofactor on the HRE, and initiate gene transcription. Because a hypoxia situation is not present, production of these genes involved in angiogenesis, cellular survival, and glucose metabolism can result in an oncogenic process. (ADRP, adipose differentiation-related protein [responsible for neutral lipid accumulation in the cell cytoplasm, resulting in the clear cell appearance]; EPO, erythropoietin; PDGF, platelet-derived growth factor; TGF, transforming growth factor; VEGF, vascular endothelial growth factor.)
In addition to VHL, other growth factors and cell adhesion pathways control HIF-1α activity.23 TGF is a ligand for the epidermal growth factor receptor (EGFR) and, upon binding, activates the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, in addition to other protein kinase pathways. Activation of the mTOR pathway increases production of HIF-1α, which can drive the oncogenic process described earlier.23,25,26 The mTOR is another pathway targeted for the therapy of RCC.
Papillary Renal Cell Carcinoma Types 1 and 2
Papillary subtypes account for 5% to 10% of RCC cases and occur in the proximal tubule. They are most commonly diagnosed as localized disease and have a more favorable prognosis than the clear cell subtype. Unlike a single mutation driving the development of clear cell disease, papillary RCC has been associated with multiple genetic abnormalities. These tumors are further subclassified into types 1 and 2. Chromophilic type 1 patients are more likely to have multiple, bilateral tumors of lower grade and have a better prognosis compared with the higher grade, singular, unilateral poor prognosis type 2 tumors.17,19,23
The majority (>80%) of hereditary papillary type 1 RCC cases are associated with germline activating mutations in the mesenchymal–epithelial transition (MET) oncogene, located at chromosome 7q31-34.27 These mutations are responsible for about 13% of sporadic type 1 disease, but chromosome 7 duplications have been found in 75% of these cases, further supporting the role of MET.23 Activation of the c-MET receptor results in increased cell proliferation and motility and decreased cellular apoptosis.26 Stabilization of HIF can also play a role in the oncogenic potential of the c-MET receptor.28
The papillary type 2 subtype occurs in patients with hereditary leiomyomatosis, which initially presents as multiple skin and uterine leiomyomas when patients are in their 20s and 30s and eventually results in formation of RCC. The associated gene for this subtype is the fumarase hydratase (FH) gene, located at chromosome 1q42.3-45. FH is a tumor suppressor gene that encodes the enzyme FH responsible for catalyzing fumarate to malate in the Krebs cycle. The gene is predominantly inactivated by loss-of-function mutations, which ultimately results in stabilization of HIF and subsequent oncogenesis.29,30
Chromophore and Oncocytoma Renal Cell Carcinoma
The chromophore and oncocytoma subtypes combined are responsible for 5% to 10% of all RCC cases and occur in the intercalated cells of the collecting system. Both are associated with a wide variety of chromosomal abnormalities, including deletions and translocations. Oncocytomas are relatively benign and rarely metastasize.31 Hereditary forms of these subtypes are associated with the Birt-Hogg-Dube (BHD) syndrome, which is characterized by hair follicle fibrofolliculomas of the face and neck and lung cysts in addition to RCC in 15% to 30% of affected individuals. The BHD gene is located on the short arm of chromosome 17 and encodes the tumor suppressor BHD gene that is responsible for the protein folliculin.17
CLINICAL PRESENTATION AND DIAGNOSIS
Imaging modalities, such as computed tomography (CT) scans, are widely used in the medical work-up of numerous conditions. ![]() As a result, most cases of RCC are now diagnosed incidentally after radiographic imaging for unrelated reasons compared with 10% in 1970.32 Fewer than 10% of patients currently present with the classic triad of flank pain, hematuria, and a palpable abdominal mass. Incidentally diagnosed tumors, or those diagnosed in the absence of signs and symptoms commonly associated with RCC, are usually smaller in size, of a lower stage, and more localized than those seen in patients who present with symptoms such as the classic triad. In addition to the classic triad, common presenting complaints are nonspecific signs and symptoms, including fatigue, weight loss, anemia, hypertension, fever, and lower extremity edema. Bone pain, adenopathy, and pulmonary symptoms are indicators of metastatic disease spread to the mediastinum or lung parenchyma.32
As a result, most cases of RCC are now diagnosed incidentally after radiographic imaging for unrelated reasons compared with 10% in 1970.32 Fewer than 10% of patients currently present with the classic triad of flank pain, hematuria, and a palpable abdominal mass. Incidentally diagnosed tumors, or those diagnosed in the absence of signs and symptoms commonly associated with RCC, are usually smaller in size, of a lower stage, and more localized than those seen in patients who present with symptoms such as the classic triad. In addition to the classic triad, common presenting complaints are nonspecific signs and symptoms, including fatigue, weight loss, anemia, hypertension, fever, and lower extremity edema. Bone pain, adenopathy, and pulmonary symptoms are indicators of metastatic disease spread to the mediastinum or lung parenchyma.32
As noted earlier, RCC can be either sporadic or hereditary. Several differences exist between the two etiologies in terms of patterns of development. Sporadic RCC most often presents as a single tumor affecting one kidney in a patient who is at least 60 years of age. These lesions may or may not be cystic in histology, and a family history is usually not reported. In contrast, those with a hereditary etiology more commonly present with numerous cystic tumors affecting both kidneys. These patients are more likely to be younger than age 50 years and may also have other malignancies or have a strong family history of RCC, in addition to other malignancies, including retinal angiomas, hemangioblastomas, and pheochromocytomas.19,32
CLINICAL PRESENTATION
Laboratory evaluation should include a complete blood count, serum calcium, serum creatinine, liver function tests, lactate dehydrogenase (LDH), coagulation profile, and urinalysis. Imaging studies, including contrast and noncontrast CT or magnetic resonance imaging (MRI) of the chest, abdomen, and pelvis, are also performed to further characterize the renal tumor, assess involvement of the inferior vena cava, and determine the patient’s disease stage. Fine-needle biopsy is used only in rare selected cases.
STAGING AND PROGNOSIS
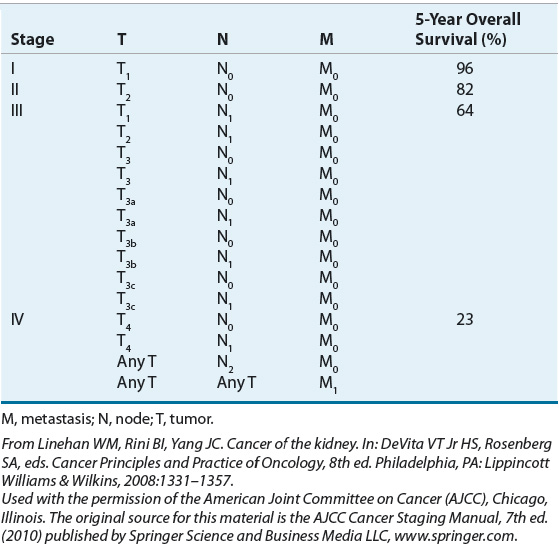
The factors associated with prognosis are positive margins after surgery, evidence of metastatic spread, presence of sarcomatoid architecture, tumor subtype, tumor grade, and tumor stage, with the latter being the most powerful prognostic indicator.33 The Union Internationale Contre le Cancer/International Union Against Cancer (UICC) and the American Joint Committee for Cancer Staging and End Results Reporting (AJCC) introduced the tumor–nodes–metastasis (TNM) staging system for RCC in 1978. The 7th edition was published most recently in 2010.34,35 The AJCC staging classification considers tumor size, number of lymph nodes involved, and the presence or absence of distant metastases. Subdivisions in the tumor (T) classification further describe the structures of the kidney that have been invaded by the tumor, including the adrenal gland, Gerota’s fascia (the layer of connective tissue surrounding the kidneys), and perinephric fat that lies between the fascia and renal capsule.35 Table 115-1 summarizes the AJCC TNM staging definitions, and Table 115-2 shows the TNM stage and corresponding 5-year OS rates.
TABLE 115-1 American Joint Committee for Cancer Staging and End Results Reporting Sixth Edition Staging
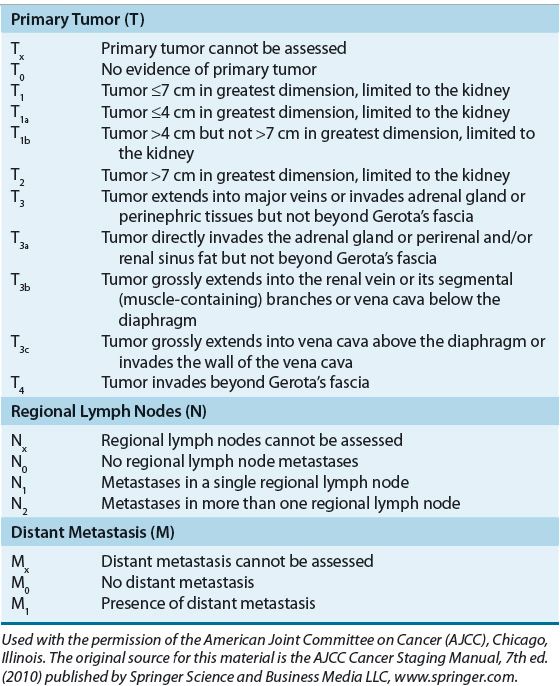
TABLE 115-2 American Joint Committee for Cancer Staging and End Results Reporting Stage Grouping
![]() In patients with metastatic disease, the Memorial Sloan-Kettering Cancer Center (MSKCC) Prognostic Factors Model was developed from a retrospective analysis of 670 patients with advanced RCC from 24 different trials at MSKCC between 1975 and 1996. The model identified five factors associated with poor prognosis: Karnofsky performance status (KPS), LDH, hemoglobin, corrected serum calcium, and nephrectomy status (later interchanged with duration of time from diagnosis to initial treatment). Patients with none of the poor prognostic risk factors are considered low risk, one or two factors are intermediate risk, and three or more factors are high risk (Table 115-3). In this analysis, 25% of patients were classified as low risk and had a median OS of 20 months, 53% were intermediate risk with a median OS of 10 months, and 22% were high risk with a median OS of 4 months. Three-year OS for the low-, intermediate-, and high-risk groups was 31%, 7%, and 0%, respectively.36 This model has been validated externally and can be used to predict survival outcomes for patients treated with IFN-based therapy.37 Another retrospective study in 353 patients with untreated RCC confirmed the MSKCC model but identified two additional independent prognostic factors: prior radiation and number of metastatic sites (none or one compared with two or more). Low risk was defined as the presence of no or one risk factor, intermediate risk as two risk factors, and high risk as the presence of three or more risk factors. Based on these criteria, 37% of the patients were classified as low risk (median OS, 26 months), 35% as intermediate risk (median OS, 14.4 months), and 28% as high risk (median OS, 7.3 months).38 In an updated analysis of metastatic RCC patients treated with targeted agents, the MSKCC model confirmed the importance of hemoglobin, corrected serum calcium, KPS, and time from diagnosis to treatment as prognostic factors for OS. Elevated neutrophil and platelet counts were also independent survival prognostic factors. Of the 586 evaluable patients treated with targeted agents (sunitinib, sorafenib, or bevacizumab), 23% were low risk with an OS that was not reached after a median follow up of 24.5 months, 51% were intermediate risk with a median OS of 27 months, and 26% were high risk with a median OS of 8.8 months. Corresponding 2-year OS rates for the low-, intermediate-, and high-risk groups were 75%, 53%, and 7%, respectively.39
In patients with metastatic disease, the Memorial Sloan-Kettering Cancer Center (MSKCC) Prognostic Factors Model was developed from a retrospective analysis of 670 patients with advanced RCC from 24 different trials at MSKCC between 1975 and 1996. The model identified five factors associated with poor prognosis: Karnofsky performance status (KPS), LDH, hemoglobin, corrected serum calcium, and nephrectomy status (later interchanged with duration of time from diagnosis to initial treatment). Patients with none of the poor prognostic risk factors are considered low risk, one or two factors are intermediate risk, and three or more factors are high risk (Table 115-3). In this analysis, 25% of patients were classified as low risk and had a median OS of 20 months, 53% were intermediate risk with a median OS of 10 months, and 22% were high risk with a median OS of 4 months. Three-year OS for the low-, intermediate-, and high-risk groups was 31%, 7%, and 0%, respectively.36 This model has been validated externally and can be used to predict survival outcomes for patients treated with IFN-based therapy.37 Another retrospective study in 353 patients with untreated RCC confirmed the MSKCC model but identified two additional independent prognostic factors: prior radiation and number of metastatic sites (none or one compared with two or more). Low risk was defined as the presence of no or one risk factor, intermediate risk as two risk factors, and high risk as the presence of three or more risk factors. Based on these criteria, 37% of the patients were classified as low risk (median OS, 26 months), 35% as intermediate risk (median OS, 14.4 months), and 28% as high risk (median OS, 7.3 months).38 In an updated analysis of metastatic RCC patients treated with targeted agents, the MSKCC model confirmed the importance of hemoglobin, corrected serum calcium, KPS, and time from diagnosis to treatment as prognostic factors for OS. Elevated neutrophil and platelet counts were also independent survival prognostic factors. Of the 586 evaluable patients treated with targeted agents (sunitinib, sorafenib, or bevacizumab), 23% were low risk with an OS that was not reached after a median follow up of 24.5 months, 51% were intermediate risk with a median OS of 27 months, and 26% were high risk with a median OS of 8.8 months. Corresponding 2-year OS rates for the low-, intermediate-, and high-risk groups were 75%, 53%, and 7%, respectively.39
TABLE 115-3 Memorial Sloan-Kettering Cancer Center Poor Prognostic Factors

Clinical Controversy…
< div class='tao-gold-member'>



