CHAPTER 4 Regulation of hematopoiesis
Introduction
Hematopoiesis, including both myelopoiesis and lymphopoiesis, is maintained throughout life by hematopoietic stem cells (HSC). Hematopoietic cells can first be detected in the yolk sac,1 followed by the aorta-gonad-mesonephros (AGM) region of the embryo proper.2,3 HSC then migrate from the AGM region to the fetal liver,4 where they undergo extensive self-renewal to generate a sufficiently large pool of HSC to sustain hematopoiesis throughout adult life.5 Prior to birth HSC seed the bone marrow (BM), and the BM becomes the primary site of hematopoiesis throughout adult life.6 To ascertain that sufficient mature blood cells are generated, hematopoietic cell production is a highly regulated process in which the majority of HSC remain quiescent under steady-state conditions, but may be induced to proliferate under conditions of stress. By contrast, the first descendants from HSC, hematopoietic progenitor cells (HPC), proliferate extensively prior to maturing to terminally differentiated cells. Although significant insights have been gained in the processes that regulate self-renewal and differentiation of HSC, which will be reviewed here, these processes remain incompletely understood. This chapter will highlight a number of the processes that are responsible for coordinating self-renewal and differentiation of HSC and HPC to ensure the controlled generation of mature blood elements under steady-state conditions and conditions of stress. Although we do address mechanisms underlying leukemogenesis or syndromes of hematopoietic failure, it follows that if any of the delicately controlled processes that will be discussed fail, insufficient or excess cells can be produced leading to these disease states.
Characteristics of hematopoietic stem and progenitor cells
The minimal definition of an HSC is a cell capable of extensive self-renewal as well as generation of both myeloid and lymphoid progeny. The concept of the hematopoietic stem cell was first proposed in the 1950s after researchers discovered that lethally irradiated mice could be rescued from BM aplasia by transplanting cells from healthy mouse BM or spleen.7 Subsequent studies demonstrated that HSC possess the unique property to self-renew and maintain the hematopoietic system throughout life. The HSC is capable of generating all blood cell lineages. During postnatal life, HSC reside in the BM where they constitute less than 0.01% of the total cell population.8 Many studies over the last two to three decades have developed tools to characterize HSC and their descendants. This has led to the development of a hematopoietic cell hierarchy wherein HSC are at the top of the hierarchy generating progressively more lineage committed cells which coincides with decreased self-renewal and proliferative potential.
HSC are the only cells that reconstitute hematopoiesis long term, and proliferate rarely in vivo. Studies using BrdU labeling in mouse HSC estimate the frequency of HSC division to be somewhere around once every month;9 however, studies using biotin label or histon 2B-GFP transgenic mice have suggested that multiple populations of HSC exist with different division kinetics and that slow dividing HSC only very rarely exist. The division rate of these different populations of HSC ranges between 0.5% per day and once every 145 days.10–12 In larger mammals, felines and non-human primates, both retroviral marking and telomere length have been used to evaluate frequency of HSC division.13,14 HSC divide in felines about once every 8 weeks,13 in non-human primates once every 25–35 weeks,14 and in humans once every year.15 Hematopoietic progenitor cells (HPC), by contrast, cannot reconstitute the hematopoietic system for the life of the recipient. However, in contrast to HSC, HPC proliferate actively to generate the millions of hematopoietic cells generated daily. At the bottom of the hierarchy are the terminally differentiated hematopoietic cells.
Phenotypic characterization
Although HSC in the mouse have been enriched to 100% purity,16 the exact phenotype of human HSC is not known, because of lack of accurate functional assays that allow enumeration of human HSC. The CD34+ population of hematopoietic cells has been shown to possess the majority of hematopoietic repopulating activity in humans, and is known to enrich for hematopoietic progenitors.17–19 Primitive human progenitors that can initiate long-term cultures or can repopulate immunodeficient animals are lineage−, CD34+, CD133+, CD38−, HLA-DRlow, c-Kit+ and Thy1low, and Lin−CD34+CD38− cells contain approximately 0.1% primitive progenitors that can repopulate the hematopoietic system of severe combined immunodeficient (SCID) mice (SCID-repopulating cells (SRC)).17,20–23 Although HPC are also CD34+, they co-express CD38, as well as cell surface proteins associated to specific lineages, such as CD33 (myeloid lineage),24 CD19 and CD10 (B-lymphoid) or CD7 (NK and T-lymphoid).25–27 Recent studies have indicated that some of the cell surface antigens previously thought to be expressed only on more differentiated cells may be present on HSC, making the characterization of human HSC even more difficult. For instance, CD33 which had been thought only to be on myeloid cells is also expressed on cord blood HSC.28
As stem cells are quiescent, they are spared from cell cycle-specific cytotoxic agents such as 5-fluorouracil, a method used frequently to enrich murine BM for HSC.29,30 In addition, stem cells express functional multidrug resistance proteins, such as p-glycoprotein (MDR1),31 and breast cancer related protein (BCRP),32 which extrude toxins from the cell. This allows selection of stem cells based on their ability to, for instance, extrude the dyes Rhodamine or Hoechst 33342.31,33 Combining these functional characteristics of HSC to cell-surface markers further enriches for human HSC, and 1/30 RholoLin−CD34+CD38− cells are SRC.34
Functional characterization
Even if we now can enrich for HSC using fluorescent activated cell sorting procedures, identification of HSC continues to depend on assays that measure stem cell function. Committed HPC can be assessed using colony-forming assays, where colony forming unit (CFU)- granulocyte-macrophage (GM), burst-forming-unit (BFU)-E, CFU-Mix can be enumerated. An additional primitive HPC subset is the high proliferative potential colony-forming cells (HPP-CFC).35 Even though HPP-CFC generate visible myeloid cell colonies and can be replated to generate new HPP-CFC, demonstrating their extensive self-renewal ability, they do not correspond to HSC.
Dexter and colleagues demonstrated in the late 1970s that long-term hematopoiesis could be established in vitro, by plating BM cells in the presence of fetal calf and horse serum. They demonstrated that this leads to the establishment of an adherent feeder of stromal cells, where hematopoietic progenitors proliferate for several weeks while generating more mature progeny.36 Subsequent adaptations of this culture system, wherein hematopoietic supportive stromal feeders are first established, whereupon hematopoietic cells can be seeded, has allowed investigators to quantify primitive hematopoietic progenitors, also termed long-term culture initiating cells (LTC-IC). LTC-IC can generate more committed CFC in a sustained manner (5 to more than 20 weeks).37,38 Although there is evidence in mice that the number of LTC-IC may correlate with repopulating HSC as progeny are only of the myeloid lineage, this assay cannot assess the frequency of true HSC.39 In vitro assays have also been developed to assess the lymphoid potential of human primitive progenitor cells, all of which also require specific microenvironments (BM stroma or stromal cell lines for B-lymphocytes, NK and dendritic cell differentiation, and either thymus derived feeders or other feeders engineered to express Notch ligands for T-cell differentiation).40–44 As is true for the LTC-IC assays described above, only lymphoid differentiation can be assessed in the latter assays, and thus again not true HSC activity. To assess the ability of cells to generate both myeloid and lymphoid progeny, ‘switch’ cultures have been developed in which the ability of single cells to give rise to both myeloid and lymphoid long-term culture initiating cells can be tested.43,45,46 Enumeration of the frequency of single cells that have the ability to generate both myeloid and lymphoid progeny comes close to assessment of HSC; however, it cannot address homing and engraftment, nor the true long-term expansion ability of cells.
In mice, HSC can be assessed by transplantation into irradiated animals. When this is done in competition with a known source of repopulating cells, the ability of putative HSC to compete with other HSC can be assessed, and when this is combined with limiting dilution analyses, the absolute frequency of repopulating cells can be measured.47–50 In general engraftment is evaluated at 4 months following transplantation; it is, however, clear that the cells generating progeny for only 4 months in vivo may not represent long-term repopulating (LTR-)HSC. Therefore, some groups evaluate engraftment at 8–10 months after transplantation to demonstrate presence of LTR-HSC,51 whereas others perform secondary transplantations to allow assessment of self-renewal ability of HSC.52,53 The development of xenogeneic transplant models in immuno-incompetent animals (immunodeficient mice such as severe combined immunodeficient (SCID) mice,54 non-obese diabetic (NOD)-SCID mice22 or NOD-SCID mice also lacking the gamma-c receptor (γc−/−),55 beige-nude-SCID (BNX) mice56 and Rag2−/−γc−/−,57 or preimmune fetal lambs58) has provided in vivo models that allow not only demonstration of multilineage differentiation but also self-renewal and repopulating ability of human cells. As human HSC have to repopulate a xenogeneic microenvironment, which may support homing, growth and differentiation of human HSC with decreased efficiency compared with a syngeneic human microenvironment, it remains to be proven that these assays enumerate all human HSC. Researchers are therefore trying to further improve mouse models for human HSC transplantation by for instance humanizing certain growth factors that poorly cross-react with human cells and/or HLA antigens to increase the efficiency of human cells to repopulate xenogeneic animal models and develop into a fully competent hematopoietic system.59 Finally, testing of the effect of certain manipulations on stem cells can also be done in large animals including non-human primate or canine models.60–63
Hematopoietic stem cell fate decisions: symmetrical vs. asymmetrical
That asymmetrical vs. symmetrical divisions occur in stem cell compartments has most elegantly been demonstrated in the model organisms such as Caenorhabditis elegans and Drosophila. One example is the fate of Drosophila germ stem cells (GSC). In the Drosophila testes, approximately 12 non-dividing somatic hub cells, located at the apical tip, make up the niche to which 5–9 GSC are attached in a characteristic rosette pattern.64 When GSC divide, one spindle pole associates with the GSC-niche interface.65 The daughter cell that remains attached to the hub cell continues to have stem cell properties, whereas the second cell, no longer attached to the hub, differentiates. The location of the GSC in Drosophila directs the fate of the cells, which has been shown to depend in part on Notch, TGFβ, and Jak/Stat signaling.
It has been shown in both C. Elegans and Drosophila that the plane of the mitotic spindle also appears to predict which daughter cell remains a progenitor/stem cell and which daughter cell differentiates. As the exact niche for HSC is not known, in vivo studies related to symmetrical and asymmetrical divisions of HSC depend on in vitro studies. These studies have shown that when primitive hematopoietic progenitor cells are cultured in vitro, they divide asymmetrically yielding one daughter cell with characteristics of the original cell and the other daughter cell having more differentiated characteristics.66 There is also preliminary evidence that a number of molecules, such as CD53, CD62L/L-selectin, CD63/lamp-3, and CD71/transferrin receptor, distribute asymmetrically which may govern the fate decisions.67
Early during development, HSC undergo symmetrical self-renewing cell divisions to generate the pool of stem cells required throughout adult life. This occurs between e14 and e18 of fetal liver development in mice. It is believed that characteristics intrinsic to the HSC as well as factors provided by the fetal liver (FL) niche must be responsible for the symmetrical divisions of HSC, and hence the net increase in HSC during this period of development. Bowie et al. demonstrated that HSC in the fetal liver are indeed significantly less quiescent than those found early postnatally.68–70 They also demonstrated that this can be explained by a number of cell intrinsic differences between FL HSC and BM HSC, including expression of some but not all of the known transcription factors and cell cycle proteins known to be involved in self-renewal of HSC, as well as differences in response to exogenous cytokines, including SCF and CXCL12. Whether the nature of the cell extrinsic signals in fetal liver stem cell niches differ from those in postnatal BM niches, to favor expansion of HSC, is not yet known but deserves further study as this may aid in developing strategies that allow HSC expansion, even postnatally or in vitro. During postnatal life HSC self-renewal occurs rarely and in an asymmetric fashion, yielding one new HSC and a cell that partakes in extensive proliferation in the transient amplifying pool to generate all mature cells. It is believed that only under stress conditions, HSC may divide symmetrically either yielding two new HSC to recreate the pool of HSC or giving rise to two differentiating cells.71
The hematopoietic stem cell niche
The notion that HSC reside in microenvironments or niches that regulate their behavior (cell quiescence vs. symmetric divisions vs. asymmetric divisions vs. differentiation) was put forward first by Schofield in 1978,72,73 even though it was not until recently that the nature of these niches has become elucidated. The bone marrow microenvironment in which HSC reside in postnatal life consists of both hematopoietic and ‘stromal’ cells.74,75 These stromal cells include endothelial cells, fibroblasts, myocytes, adipocytes and osteoblasts. Stromal cells produce and deposit a complex extracellular matrix (ECM) and produce hematopoietic cytokines that induce or inhibit progenitor proliferation and differentiation.76,77 Hematopoietic cells interact through cell-surface receptors with either immobilized or secreted cytokines, with adhesive ligands present on stromal cells or ECM components, and other hematopoietic cells. The combined effect of cell–cytokine, cell–cell and cell–ECM interactions governs the normal hematopoietic process.
The fact that one can now identify murine HSC based on cell surface antigens to near homogeneity has allowed investigators to determine that HSC can be found either in close proximity with endosteal osteoblasts (the osteoblastic niche)78,79 or near small blood vessels (the vascular niche)16 (Figure 4.1). The osteoblastic niche may be providing a favorable environment to maintain the HSC in a quiescent state, whereas the vascular niche may be providing signals for differentiation and mobilization to the peripheral blood.
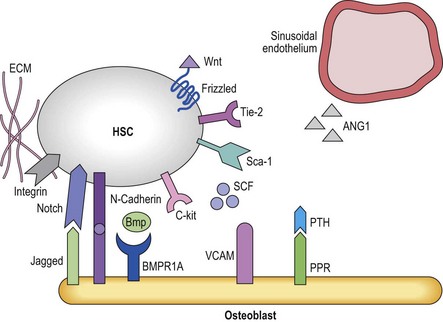
Osteoblastic niche
The development of bone has long been linked to the formation and function of the bone marrow.80 A direct role in vivo has more recently been established by a number of studies. In the adult mouse, HSC reside near the endosteum lining the BM cavity.78,79 The finding that osteoblasts play a role in HSC homeostasis comes from studies in which the osteoblast compartment of the bone was expanded by overexpression of parathyroid hormone (Pth) and its receptors,81 and a second series of studies in which the BMP-receptor-1a was knocked out, both leading to an increase in HSC.82 In both studies an increase in osteoblasts correlated with an increase in HSC.81 Osteoblasts and HSC were found to interact through N-cadherin interactions, although the role of this interaction is unclear.81 As these genetic manipulations increase HSC numbers,83 it is believed that the osteoblastic niche regulates HSC maintenance and self-renewal, perhaps by direct cell–cell interactions via the Notch84 and N-cadherin pathways.85 There is evidence that multiple receptor ligand interactions may affect HSC localized in the osteoblastic niche; many of these will be discussed later in this chapter. They include the morphogens BMP4 and Wnt3a,86,87 interactions between Notch on HSC and Jagged-1 in the niche,88 Tie2 expressed on HSC and angiopoietin-1;83 c-Kit and c-Kit-L;89 or β1 integrins that interact with both VCAM-190 and osteopontin.91,92 Osteoblasts also appear to affect adjacent cells including osteoclasts93 that then affect HSC fate decisions. Likewise, there is evidence that RANK-L produced by osteoclast mediated breakdown of the bone also influences HSC behavior.94 However, other studies demonstrated that ablation of osteoblasts using a Collagen-1a1 conditional knockout mouse leads first to the depletion of more committed progenitors and later to depletion of the HSC compartment, which contradicts the notion that osteoblasts would preserve the HSC pool.95 Like murine HSC, human primitive progenitors can be found in close proximity with the endosteal lining of the marrow cavity,6 although the molecular mechanisms underlying these interactions and the role of endosteal cells in human HSC regulation are unknown.
Vascular niche
In vivo, endothelial cells line the sinusoids of the BM cavity. As in other tissues, entry and exit from the marrow requires that HSC/HPC and mature blood cells pass through the endothelial barrier of the sinusoids. Unlike most sinusoids, BM sinusoids only have a single layer of endothelial cells, which allows for increased permeability.96 Aside from serving as a gatekeeper for cell entry and exit from the BM, it is believed that BM endothelial cells also play a role in regulating hematopoiesis. Aside from endothelial cells, additional cells in the vicinity which may regulate HSC in vivo include pericytes (thought to be the in vivo mesenchymal progenitor cell), megakaryocytes and perivascular reticular cells. Although identification of murine HSC via the SLAM receptors has shown that HSC reside near endothelial cells,16 the exact contribution of the different cell types in the vascular niche to HSC proliferation and differentiation control are, however, less well understood.
Intrinsic regulation of hematopoiesis
HSC behavior is controlled in part by factors exogenous to the HSC, but also in part by cell intrinsic mechanisms. The latter is the result of a complement of transcription factors (TFs) expressed specifically in HSC but not HPC, which can bind to the promoter regions of specific target genes to allow their transcription and ultimate production of the proteins typical for HSC and not HPC. TFs also recruit co-factors that can modify the chromatin surrounding target genes, to allow easier or more difficult transcription of certain regions of the genome, which is part of the epigenetic regulation of gene expression. The latter consist of methylation and acetylation of histones, proteins to which the DNA is bound, as well as of methylation of CpG islands in the promoter regions of target genes, which prevent binding of TFs to allow transcription. The role of these basic processes, important in all steps of development to allow orderly process of lineage commitment and specification, in hematopoiesis has recently been extensively reviewed by Rice et al., and we refer readers to that exhaustive review for more detailed information.97
For many of these genes, aberrant expression is associated with abnormal hematopoiesis, in many instances leukemia development. For instance, recurrent chromosomal translocations in human leukemias that deregulate expression and/or function of genes such as TAL1, LMO2, NOTCH1, and HOX genes98–102 result in aberrant proliferation of HSC/HPC without maturation, findings that have led to the identification of these genes as key regulators of hematopoiesis.
Hematopoietic stem and progenitor cell specific genes
A number of genes have been identified that are indispensable for the creation of HSC and their lineage specific progeny during development. These include, among others, LMO2, TAL1, GATA2, GATA1 and PU.1.103–107 LMO2, TAL1 and GATA2 are expressed already in the hemangioblast, or the mesodermal cell that can generate both endothelium and hematopoietic cells.104–110 Loss of any of these three genes inhibits the emergence of HSC during development. Subsequent commitment to lineage specific hematopoietic cells such as erythroid vs. myeloid and B-cells is then governed by the lineage specific expression of GATA1 and PU.1, respectively.111
Homeobox genes
This is a family of highly preserved genes containing a DNA-binding domain, termed homeodomain, that play important roles in many aspects of development, including normal as well as malignant hematopoiesis.112–117 Aside from the typical HOX genes, there is a second family of homeodomain-containing genes, the three amino acid loop extension (TALE) family of transcription factors, which include PBX1 and MEIS1.118,119 HOX proteins interact with PBX1 to form a complex with MEIS1, and regulate gene expression.120–122 Mice deficient in either PBX1 or MEIS1 are embryonic lethal.123,124 MEIS1 may play a role in expansion of fetal liver HSC as MEIS1−/− fetal liver cells have impaired competitive repopulation abilities.124,125
The HOX family consists of four clusters (A–D) which are located on different chromosomes. Within each cluster, HOX genes are further subclassified from the 3′ to 5′ end in 13 paralog groups, based on sequence homology. Different HOX genes are expressed in HSC and HPC, with HSC being characterized by presence of HOXB3, HOXB4 and HOXA4,125,126 whereas HOXA9 is more highly expressed in HPC that will give rise to granulopoiesis and T- and B-cell lymphopoiesis.113 Although these genes are very important regulators of hematopoiesis, loss of a single homeobox gene does not always lead to hematopoietic defects due to redundancy between different HOX genes. For instance loss of HOXB4 or HOXB3 alone does not lead to defective hematopoiesis, and even combined loss of HOXB4 and HOXB3 only results in a moderate decrease in hematopoietic cell output. By contrast, aberrant expression of HOX genes has been associated with different types of leukemia, including HOXA9 and HOXC13, pointing to their role in HSC and HPC self-renewal and differentiation. When the HSC specific HOXB4 gene is force expressed in murine and human HSC, significant increased proliferation of cells with HSC characteristics in vitro is observed. When grafted in competitive repopulation assays in vivo, HOXB4 transduced cells out-compete normal HSC, without significant skewing of hematopoiesis or frank leukemia development.112,117,127 As increased self-renewal of HSC may also be possible by simple HOXB4 protein transfection, this strategy is being contemplated to induce HSC expansion clinically.128 Kyba et al. also demonstrated that forced expression of HOXB4 in murine embryonic stem cell (ESC) aids in the generation and engraftment of competent HSC from ESC, possibly by inducing the expression of the chemokine receptor CXCR4.129,130 Although HOXB4 does not seem to promote leukemia, deregulation of other HOX family members is linked to hematopoietic malignancies such as leukemia. A number of HOX genes are found fused with NUP98 in human leukemia,131,132 and expression of HOXA9 is associated with a poor prognosis in patients with acute myelogenous leukemia.
Polycomb genes
Upstream regulators of HOX genes are among others the mixed-lineage leukemia (MLL), a common target of chromosomal translocations in human acute leukemias, which induces gene expression during development and the Polycomb gene (PcG) family, responsible for suppression of gene expression. Both of these are believed to regulate gene expression by complex epigenetic mechanisms. The Polycomb group (PcG) represents a gene family of transcriptional repressors first identified in Drosophila. They play a key role in many developmental processes by regulating self-renewal and proliferation, senescence and cell death,133–137 and this via interactions with the initiation transcription machinery138,139 as well as chromatin-condensation proteins and histones.140,141 The PcG proteins are organized in Polycomb regulatory complexes (PRC). Two PRC complexes have been identified, consisting of EZH, EED and SUZ12 (PRC2) whereas BMI1 and RAE28 are part of the PRC1 complex.142 A number of PcG genes are expressed in differentiating hematopoietic cells, whereas BMI1 and RAE28 are found highly expressed in HSC.136,143–145 The role of BMI1 in HSC has been elucidated using both knock-out studies and by forced expression in HSC. HSC from BMI1−/− mice fail to long-term repopulate lethally irradiated recipients suggesting that BMI1 plays a role in HSC self-renewal. In addition, the HSC compartment in BMI1−/− senesces significantly faster than in WT mice. Similar defects are also seen in other stem cell compartments of BMI1−/− mice.146–148 By contrast, forced expression of BMI1 enhances self-renewal in vivo.136,149,150 Similar findings were seen in RAE28−/− fetal liver HSC, which have impaired engraftment potential.144 MEL18, another PcG gene, which is expressed in a reciprocal fashion with BMI1, by contrast inhibits HSC self-renewal.151,152 There is also evidence for a role of PcG proteins part of the PRC2 complex in HSC proliferation, including EED, EZH2 and SUZ12.153–156 Aside from being upstream regulators of HOX genes, several of the PcG genes may act by modifying the expression of cell cycle regulators p16INK4a/p19ARF.151
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


