CHAPTER 35 Acquired bleeding disorders
Introduction
Acquired disorders of hemostasis are significantly more common than inherited disorders of hemostasis and as such are frequently encountered in routine clinical practice. Acquired disorders of coagulation may be physiological such as those that are seen in pregnancy, the newborn and with advancing age or they may be pathological. The latter often arise as a complication of multi-system disease and may therefore be associated with multiple clotting abnormalities (Table 35.1).
Table 35.1 Disorders associated with an acquired hemostatic defect
| Physiological deficiencies Neonates | Vitamin K deficiency Neonates Gastrointestinal disease Vitamin K antagonists Biliary obstruction Liver disease Miscellaneous, e.g. cephalosporins, parenteral nutrition |
| Liver disease | Disseminated intravascular coagulation |
| Drug induced bleeding Anticoagulants Thrombolytic agents Anti-platelet agents Miscellaneous | Renal disease and renal failure |
| Massive blood transfusion | Cardiopulmonary bypass and extracorporeal circuits |
| Acquired inhibitors of coagulation Factor VIII inhibitors Other factor inhibitors Anti-phospholipid antibodies | Miscellaneous Snake venoms and other toxic agents Myeloproliferative disorders Malignancy Paraproteinemias |
Physiological deficiencies
Neonates
The coagulation system of the newborn infant is complex and reflects hepatic immaturity. Most of the clotting factors are present in reduced concentration in the newborn infant apart from factors V, VIII and fibrinogen.1–3 These physiological deficiencies in clotting factors result in prolongation of the prothrombin time (PT) and activated partial thromboplastin time (APTT) and as a consequence of this, reference ranges reflecting both gestational and neonatal age must be used to assess coagulation in the neonate.1–3 The platelet count is normal in the neonate although there may be a qualitative platelet abnormality. Fibrinolysis in the neonate is similar to that of adults.
Drug-induced bleeding disorders
Drugs are a common cause of an acquired bleeding disorder. In many cases the drug may be obvious, e.g. an anticoagulant, but in other cases it may be less clear, as with the inhibitory effect on vitamin K metabolism observed with some cephalosporins.4
Heparin
Unfractionated heparin (UFH), the low molecular weight heparins (LMWHs) and fondaparinux (a synthetic pentasaccharide) are anticoagulants that potentiate the action of antithrombin by increasing its inhibitory activity.5 The inhibitory activity of UFH is directed against both thrombin (IIa) and factor Xa whereas that of the LMWH is primarily against factor Xa.5 Fondaparinux has exclusively anti-Xa activity. Bleeding in patients receiving heparin is usually secondary to excessive anticoagulation. Heparin is metabolized by the liver and excreted by the kidneys and LMWHs may accumulate in patients with impaired renal function6 and a dosage adjustment may be required if the creatinine clearance is less than 30 ml/min.
In patients receiving unfractionated heparin intravenously the rate of major hemorrhage ranges from 0%–7%. Fatal bleeding with a 5–14 day course of heparin ranges from 0%–2%. The risk of hemorrhage is significantly increased if there is concomitant use of other anticoagulants particularly anti-platelet agents such as aspirin or clopidogrel. Other patient-specific factors, including impaired renal function, disordered liver function, thrombocytopenia and invasive procedures, significantly increase the risk of bleeding. In individuals who are actively bleeding, unfractionated heparin can be effectively neutralized by protamine sulphate, a strongly basic drug that binds to the heparin. A dose of 1 mg of protamine sulphate will neutralize approximately 100 units of heparin. In overdose, protamine sulphate can function as an anticoagulant and no more than 50 mg of protamine sulphate should be administered at any one time. Protamine sulphate neutralizes only 60% of the anti-Xa activity of the low molecular weight heparins and is, therefore, less effective in correcting the bleeding problems associated with their use.7 Protamine sulphate does not bind to fondaparinux and is, therefore, of no value in the management of patients on fondaparinux who are bleeding.
Laboratory monitoring8
Hirudin and bivalirudin
Hirudin was originally isolated from the medicinal leech Hirudo medicinalis but now is available in a recombinant form (r-hirudin or lepirudin). Bivalirudin is a small (MW 2180 Da) synthetic peptide modeled after hirudin, which contains 20 amino acids and two thrombin-binding domains. Bivalirudin and lepirudin are direct thrombin inhibitors and bind to both the catalytic site and the anion-binding exosite of circulating and clot-bound thrombin but do not require antithrombin for their anticoagulant activity.9–11 Lepirudin and bivalirudin inhibit the conversion of fibrinogen to fibrin but also other thrombin-catalyzed reactions, for example activation of clotting factors and thrombin-induced platelet aggregation.
Laboratory monitoring
Therapeutic anticoagulation with lepirudin or bivalirudin is commonly monitored by the activated partial thromboplastin time (APTT) but there is considerable inter-individual variability in the degree of prolongation of the APTT at identical plasma levels of these drugs. The ‘ecarin’ clotting time (ECT) has been suggested as a more accurate test for monitoring individuals receiving direct thrombin inhibitors12 including dabigatran if needed. Ecarin is isolated from the venom of the saw-scaled viper Echis carinatus and in the assay a known amount of ecarin is added to the plasma. Ecarin activates prothrombin to meizothrombin – this activity is inhibited by lepirudin but is unaffected by heparin. The meizothrombin induces clotting via fibrinogen cleavage to fibrin. This prolongation in the clotting time increases in a linear fashion with increasing concentrations of lepirudin but also with bivalirudin, dabigatran and argatroban, another direct thrombin inhibitor. The ecarin chromogenic assay employs a similar approach but the concentration of meizothrombin is measured using a chromogenic substrate.13
Warfarin and vitamin K antagonists
Warfarin is a 4-hydroxycoumarin derivative that exerts its action by blocking the regeneration of vitamin K from its epoxide. The major complication of all vitamin K antagonists is bleeding and this risk increases as the intensity of treatment, i.e. the INR, increases.14,15 Independent risk factors for bleeding during long-term warfarin therapy include age greater than 65 years, a history of past gastrointestinal bleeding, stroke, atrial fibrillation and one or more of three co-morbid conditions: myocardial infarction, renal insufficiency and severe anemia.16 For any individual the risk of bleeding is related to the duration of anticoagulant therapy although the risk may be higher in the early phase of treatment. Most studies in unselected groups of patients suggest that the risk of major bleeding is ~3% per annum and that CNS hemorrhage occurs at a rate of 0.1% per annum.17
The anticoagulant action of warfarin is potentiated by many drugs and these include:
Minor bleeding episodes in patients receiving oral anticoagulants may be treated with local measures and withdrawal of the drug. In cases of severe or life-threatening hemorrhage, rapid reversal of anticoagulation is required and this is most effectively achieved by the use of a combination of vitamin K and clotting factor concentrates (containing factors II, VII, IX and X) and less effectively by vitamin K and fresh frozen plasma.18
Laboratory monitoring
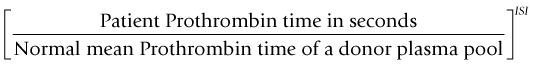
where the ISI (International Sensitivity Index) is a value derived by calibrating the tissue factor used in the assay against an international WHO standard, the ISI of which is 1.0. For an individual who is not on warfarin the INR is 1.0. The target INR for any patient varies depending upon the indication for treatment but is usually 2.5, 3.0 or 3.5. The risk of bleeding on any VKA increases as the INR increases and patients with a target INR of 3.5 have a significantly greater risk of hemorrhage than those with a target INR of 2.5.17
Thrombolytic agents
T-PA, urokinase, reteplase and tenecteplase produce their pharmacological actions by converting plasminogen to plasmin at the site of fibrin deposition. In contrast staphylokinase and streptokinase bind to free plasminogen in the plasma leading to systemic hyperfibrinolysis. Bleeding occurs in 3–40% of patients receiving thrombolytic therapy and this risk is greatly increased in patients who are also receiving anti-platelet drugs or other anticoagulants.19 Thrombolytic therapy predisposes to bleeding by depleting the plasma concentration of procoagulant proteins and by the generation of anticoagulant fibrin(ogen) degradation products. Thrombolytic therapy cannot distinguish between a pathological thrombus occluding a critical vessel, e.g. coronary artery, and a physiological thrombus that is preventing bleeding from a critical site, e.g. in the cerebral circulation. Platelet function in patients receiving thrombolytic therapy is also impaired because of inhibition of platelet aggregation by high levels of FDPs and also by impaired platelet adhesion by plasmin-induced proteolysis of glycoprotein Ib (GpIb) and von Willebrand factor (vWF).20
For patients receiving thrombolytic therapy and who develop minor bleeding episodes the thrombolytic agent, together with any anticoagulant or anti-platelet agent, must be discontinued. For life-threatening bleeding episodes, a fibrinolytic inhibitor should be given e.g. tranexamic acid. Fresh frozen plasma and/or cryoprecipitate or a fibrinogen concentrate should be given to restore depleted clotting factors.19
Laboratory monitoring
Laboratory monitoring of thrombolytic therapy is often unnecessary when its administration is short-term. However, during a more prolonged infusion (>24 hours) sequential monitoring may be of value. Fibrinolytic therapy alters most laboratory tests of coagulation but few tests predict either the efficacy of thrombolysis or the risks of bleeding. The APTT is prolonged in patients receiving thrombolytic therapy because of depletion of fibrinogen, factors V and VIII and the generation of high levels of fibrin(ogen) degradation products. An APTT ratio of 1.5 indicates significant systemic fibrinolysis.19 Plasma fibrinogen concentration falls during thrombolytic therapy reflecting the presence of free plasmin within the circulation. The fibrinolytic activity of the plasma can be measured by means of the euglobulin clot lysis time (ELT) which is shortened in patients receiving thrombolytic therapy but this is rarely, if ever, used. The thromboelastogram (TEG) may be of value and is considerably easier to perform than the ELT.
Anti-platelet drugs
Laboratory monitoring
There is increasing interest in monitoring platelet function in patients receiving anti-platelet drugs to identify those individuals who demonstrate drug ‘resistance’ and may have a reduced benefit.29–34 Platelet aggregation studies or the use of the platelet function analyzer 100 (PFA-100™) may be of value in identifying these patients.35–37
Hemostatic defects associated with vitamin K deficiency
Vitamin K and vitamin K deficiency
Vitamin K deficiency in neonates and young infants
Three types of vitamin K deficiency are seen in the newborn child and young infant:
Laboratory findings
Adults and children with vitamin K deficiency both show a normal platelet count (unless there is an associated pathology such as liver disease or DIC which may result in thrombocytopenia), a prolonged prothrombin time (PT), a prolonged activated partial thromboplastin time (APTT) but a normal thrombin time and fibrinogen level. The functional activity of the vitamin K dependent clotting factors are reduced and dysfunctional forms – PIVKAs – can be detected in the plasma of affected individuals.40
Management of vitamin K deficiency
The principles of treatment involve treating the underlying cause, the administration of vitamin K and, in cases of severe hemorrhage, transfusion with fresh frozen plasma or in some cases a plasma-derived clotting factor concentrate containing vitamin K clotting factors (II, VII, IX and X). There were some concerns that the use of parenteral vitamin K, but not oral vitamin K, in the newborn infant was associated with an increased risk of childhood cancer. However, subsequent studies have not confirmed these early findings.38,41
Hemostatic defects in liver disease
The liver is responsible for the synthesis of all the coagulation factors apart from von Willebrand factor (vWF). The liver also synthesizes either completely or in part many of the proteins involved in the regulation of coagulation – antithrombin, protein C, protein S, heparin co-factor II and those involved in fibrinolysis – plasminogen and α2-antiplasmin. The liver is also responsible for the clearance of activated clotting factors that are generated by the clotting cascade and during fibrinolysis. Liver disease is therefore associated with a major disruption of the clotting system resulting in an increased risk of hemorrhage. Factors V and VII are sensitive markers of hepatic function and may be used as an index of severity.42
Thrombocytopenia is a common finding in liver disease and is often due to sequestration of platelets within the spleen – hypersplenism. Thrombocytopenia may also be seen in association with alcohol abuse, folate deficiency, DIC and in some cases of viral hepatitis where the causative virus may have a direct effect upon megakaryopoiesis or accelerate peripheral destruction.43 A qualitative platelet abnormality is often seen in patients with liver failure which further exacerbates the bleeding tendency.
Fibrinogen is relatively well maintained in liver disease until the terminal stages when the levels may drop dramatically. In addition, as a result of an increased sialic acid content of fibrinogen, patients with liver failure may develop an acquired dysfibrinogenemia resulting in slow fibrin polymerization and a relatively unstable fibrin clot.44,45 Abnormal fibrinogens and non-carboxylated prothrombin are also synthesized by patients with primary hepatocellular carcinoma and have been used as markers of these disorders.44
Many patients with liver disease have evidence of systemic fibrinolysis secondary to reduced synthesis of α2-antiplasmin, reduced clearance of t-PA and low grade DIC.46–50 Primary hyperfibrinolysis may result in severe bleeding problems following surgery in patients with liver disease where tissue damage results in the release of large amounts of plasminogen activators which swamp the impaired protective mechanisms of the liver resulting in systemic fibrinolysis.
Chronic low-grade DIC is a common feature of liver disease. This occurs secondary to release of tissue thromboplastin from the damaged hepatocytes, reduced synthesis of the inhibitors of coagulation – antithrombin, protein C and protein S – and reduced clearance of activated clotting factors. Ascitic fluid appears to contain a potent thromboplastin-like material and may result in severe DIC following creation of a peritovenous shunt in which large amounts of ascitic fluid are infused directly into the circulation.51
Laboratory findings in liver disease
There is increasing evidence that highlights the poor correlation between bleeding and the results of various laboratory tests of hemostasis in patients with liver disease. Thrombin generation testing and measurement of platelet adhesion are normal in these patients. This has implications for managing patients about to undergo invasive procedures when historically fresh frozen plasma has been administered to these patients to reduce their perceived increased risk of bleeding based upon the results of conventional laboratory tests of hemostasis.49
Management of the coagulopathy of liver disease
Patients with liver disease may require no treatment unless they are actively bleeding or about to undergo an invasive procedure. In such cases, patients may require vitamin K, fresh frozen plasma and occasionally cryoprecipitate or fibrinogen concentrates to correct the clotting factor deficiencies and platelet transfusions to maintain the platelet count above 50 × 109/l. Patients with liver disease may develop catastrophic variceal bleeding and in such cases in addition to local measures, replacing clotting factors and platelets, rVIIa may have some benefit. Prothrombin complex concentrates have been avoided in patients with liver disease because of concerns that they could precipitate a thrombotic event.52,53 Increasingly, however, they are now being used in such patients as a rapid means for reversing the coagulopathy.
The INR in liver disease
The INR is frequently measured in patients with liver disease and serves as a prognostic factor in both the model for end-stage liver disease (MELD)54 and Child–Pugh scoring system55 and in addition to determining the prognosis, is also used to prioritize patients for transplantation. However, the INR is designed to monitor patients on warfarin and not with liver disease56 and may impact on prioritization for liver transplantation.57 For these reasons an INR using a tissue factor that has been calibrated for patients with liver disease, ‘ISILiver’, has been proposed.57,58
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


