9.1 Introduction
The finished-product specifications define the quality that every product must exhibit in order to fulfill the requirements of safety and efficacy. These critical quality aspects are identified and studied during product development and the manufacturing process.
In the application dossier for market authorization, the quality necessary in order to go to market is set down in the Module 3, “Quality Data,” part of the common technical document (CTD) (Figure 9.1). In this dossier, the qualitative and quantitative characteristics, test procedures, and acceptance limits with which the medicinal product must comply during its shelf life are detailed. The specifications are the acceptance limits applied to the tests performed on the finished product. They are a selection of the characteristics studied during pharmaceutical development and considered crucial to product quality. The characteristics of the batches of drug product used in pivotal clinical studies decide the specification limits [1].

The shelf life of the medicinal product is primarily established on the basis of the content of active constituents (efficacy), on the admissible level of breakdown products or impurities (safety), and on the consistency of pharmacotechnical properties (quality). The applicant for marketing authorization sets the specification limits at the time of batch release, such that the limits proposed at the end of the shelf life are guaranteed. These specifications at manufacture may be different from those at expiry.
An inhalation product is a solid or liquid drug formulation contained in a delivery device, intended for deposition in the respiratory tract. The inhalation product can deliver one or more drug, dissolved or dispersed in a suitable vehicle for administration, to the lungs as an aerosol. It is available as a single-dose or multidose, pressurized or nonpressurized, metered or device-metered product.
The therapeutic effectiveness of an inhalation product is dependent on the ability of the drug to be deposited at the intended site in the lungs for local and/or systemic effect. The safety and efficacy of an inhalation product is governed by quality attributes of the drug product, as well as the administration appropriateness and physiological and anatomical characteristics of the user’s lungs. With reference to inhalation products involving metering, the dose delivered, the number of activations to provide the recommended dose, and the total number of activations per inhaler must be indicated on the label to ensure a safe, high-quality, and efficacious treatment is provided.
The medicine agencies dictate the specification tests and acceptance limits necessary for an inhalation product intended for market [2]. In Europe, this responsibility is allocated to the European Medicine Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP), following consultation with the competent authorities of European Union (EU) member states and Health Canada. The specifications are communicated as scientific guidelines, incorporating the monograph information of the European Pharmacopoeia or equivalent. The guidelines harmonize the interpretation and validation of quality, safety, and efficacy aspects of inhalation products. They have been introduced with flexibility due to the range of inhalation products, which have considerable disparity in formulation and delivery-device characteristics.
9.2 Inhalation-Product Specifications
Pressurized metered-dose inhalers (pMDIs), device-metered or premetered dry powder inhalers (DPIs), single-dose or multidose nebulization products, and metered-dose nebulizers (MDNs), their manufacturing processes, container closure systems, drugs, and excipients, are all subjected to quality assessment.
Table 9.1 lists the specification tests for inhalation products in accordance with their dose and particulate, chemical, and microbiological properties. The listed tests are a combination of EU [2] and additional arbitrary selected US Food and Drug Administration (FDA) [3, 4] requirements.
Table 9.1 Specification tests for inhalation products. Tests in grey are those listed in [2]. + test requested; − test avoidable. © EMEA 2006 http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003568.pdf [Accessed 12th September 2012]
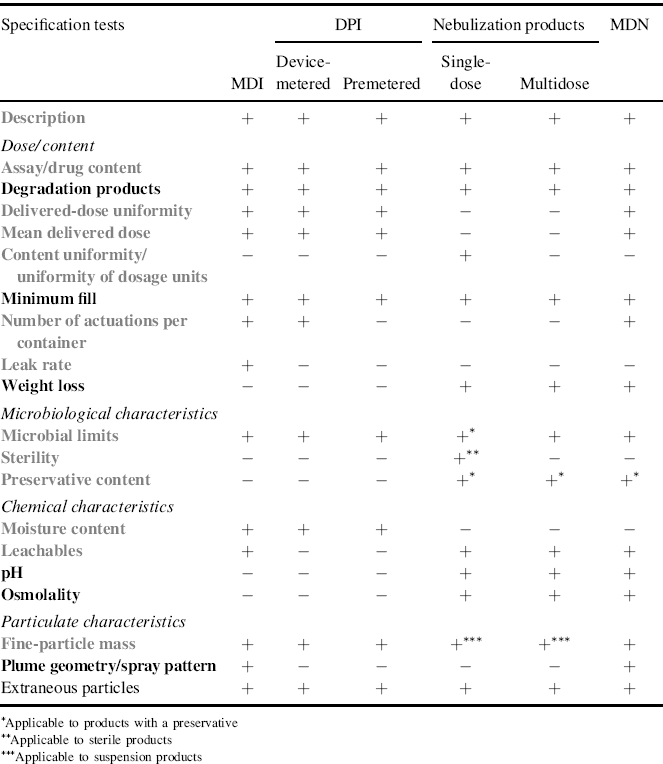
The quality attributes of an inhalation product must be maintained within approved limits from the date of batch release till the end of shelf life. As such, storage and handling conditions are based on the need to maintain the intended quality. The common tests required to qualify an inhalation product are description, dose/content, microbiological characteristics, chemical characteristics, and particulate characteristics (Table 9.1).
9.2.1 Description
A description of the formulation and the delivery device is essential to inhalation product recognition and as an indicator of integrity. The pharmacological effects of the drug and the excipient functions are also described. The description encompasses the composition and the appearance of the formulation and delivery device, which includes color, clarity, size, and shape. The actuator, immediate packaging material, and other parts which can influence drug delivery have to be described. In the case of products for nebulization, the immediate packaging is the container closure system, which for nebules (monodose solution) is the translucent low-density polyethylene ampoule.
9.2.2 Identification
The identity of the drug in an inhalation product is assessed with regard to a reference standard by use of techniques such as chromatography with ultraviolet or infrared spectroscopy detection. Complementary chromatographic procedures with a single integrated step, such as high-performance liquid or gas chromatography mass spectrometry, are used as well. The counter ion of a drug salt should also be identified. The identification methods should be specific in the case of a chiral drug.
9.2.3 Drug Content
The amount of drug substance in the entire container is determined using a stability-indicating analytical method. The common assay limits of drug content for a medicinal product are ±5% of the drug in the label at the time of batch release.
The drug content can be analyzed using high-performance liquid chromatography or another assay procedure which allows the detection of degraded drug and changes in drug concentration in the formulation. A chiral assay is used to demonstrate that there is an insignificant racemization of chiral drug during manufacture and storage.
The drug content of a multidose inhalation product is expressed as the amount of drug per weight or volume unit. The drug content of a single-dose solution for nebulization is defined as the amount of drug per dosage unit [5].
9.2.4 Impurities and Degradation Products
Drug degradation products are impurities due to chemical changes during manufacture or storage, under the influence of light, temperature, or pH, or by reaction with the excipient and immediate container closure system.
The contents of a degradation product can be determined using the drug-content assay method, referring to the drug content in the product. Alternatively, they can be examined versus reference standards if the identity of the degradation product is known.
Both identified and unidentified impurities found in a threshold concentration of ≥0.1% must be specified. Acceptance limits should be set for individual and total impurities. A stricter specification threshold may be adopted with an inhalation product administered at high daily doses, since the degradation products in a larger dose can potentially affect the therapeutic status of the medicine [2–4].
9.2.5 Preservative Content
Those solutions intended for multiple administrations are added with preservatives in order to restrict microbial growth. This control is needed for nebulization products and MDNs which contain a preservative, a type of excipient, unless the preparation has an antimicrobial property. Preservatives are not required in dry inhalation products, due to the limited propensity for microbial growth in solid product. The preservative content of nebulization solutions and MDNs is analyzed using specific techniques. The acceptance criteria for product content should be based on the levels of preservative necessary to maintain the microbiological quality of the product.
9.2.6 Microbial Limits
The presence of microorganisms in nonsterile products poses the threat of reducing or inactivating product activity, affecting the health of the user. Microbial examination of inhalation products is required in order to restrict the bio-burden of the dosage form. For inhalation products, the acceptance limits, expressed as colony forming unit (CFU)/g or CFU/mL, are 102 for total aerobic microbial count and 101 for total combined yeast/mold count. Staphylococcus aureus, Pseudomonas aeruginosa, and bile-tolerant gram-negative bacteria should be absent.
A microbial limit test is conducted through the incubation of a product sample in an appropriate agar or broth medium at specified temperatures and for specified durations, according to membrane-filtration, plate-count, surface-spread, or most-probable-number preparation protocols.
Casein soya bean digest agar/broth and Sabouraud-dextrose agar are the media employed in total aerobic microbial count and total combined yeast/mold count determination, respectively. Selective agar is used in the detection of specific microorganisms. In all microbiological examinations, a neutralizing agent may be added to remove the activity if antimicrobial agents are present [6, 7].
9.2.7 Sterility
It is advocated that single-dose nebulization products be prepared in sterile conditions in order to comply with the sterility test (no evidence of microbial growth upon incubation of a product sample in suitable culture media for 14 days).
Fluid thioglycolate and Casein soya bean digest media are primarily used in culture of anaerobic bacteria, aerobic bacteria, and fungi. The product sample is introduced into culture medium following membrane filtration or through direct inoculation.
The probability of detecting microbes via sterility test increases with product sample number and the readiness of growth of any microorganisms present. The sterility test is a destructive assay and only the selected samples are tested. An appropriate sampling plan should therefore be adopted, considering batch size, volume of preparation per container, sterilization method, and so on. In the case of aseptic production, it is recommended that samples manufactured at the beginning and the end of the batch be selected, as well as samples made after significant manufacturing interventions [8].
9.2.8 Delivered-Dose Uniformity
The delivered-dose uniformity test is conducted according to a pharmacopoeia method, or a suitable validated alternative. Limits applied should be consistent with the pharmacopoeia, testing both intra- and interdevice variability. The mean delivered dose and delivered-dose uniformity of products for nebulization need not be assessed.
It is however essential to characterize the uniformity of doses delivered from metered inhalation products, since the reproducibility of doses is affected by the actuation procedures.
The apparatus set-up for dose collection incorporates the sequential assembly of inhaler, mouthpiece adapter, sample collection body housing a filter, and vacuum pump connection (Figure 9.2, from right to left). The aspiration airflow rate created by a vacuum pump can be 28.3 L/min or higher.
Figure 9.2 Set-up of a dose-collection apparatus with an MDI in place (from left to right: vacuum pump connection, filter, sample collector, adaptor, MDI)

A total of 10 doses discharged in the sample-collection body are assessed. The doses have to be collected over the life span of the inhaler—that is, at the beginning, middle, and end of the dose number stated on the label. The drug recovered from the dose actuated into the collection body is assayed. The delivered-dose-uniformity test must be conducted for each individual drug if the inhalation product contains more than one drug, and under breath actuation conditions if the inhaler is breath-operated.
The uniformity of delivered doses must comply with ±25% of the average dose for 9 out of 10 results. All should comply with ±35% of the average dose. If two or three values fall outside the range 75–125%, further tests on two more inhalers are required. In the latter, the uniformity of delivered doses must comply with ±25% of the average dose for 27 out of 30 results, and all should comply with ±35% of the average dose.
The delivered-dose uniformity of a solution formulation may be represented by uniformity of weight per actuation instead of uniformity of drug content of delivered doses. Justification of the reproducibility of drug content in doses delivered is requested in this case.
The uniformity of delivered doses among the containers of a batch must be assessed in addition to those from a single container. The sample size is 10 containers. The uniformity of dose per actuation between containers follows the same acceptance criteria applied to delivered-dose uniformity for a single container. A failed test should be repeated with an additional 20 containers [9].
9.2.9 Content Uniformity/Uniformity of Dosage Units
This test applies to solutions for nebulization packaged in a single-dose container. In the case of device-metered DPIs, MDIs, and MDNs, the dose-content uniformity is monitored using the previously described delivered-dose-uniformity assessment. Content uniformity should be investigated in samples removed from the containers as per the instructions provided to users and health care professionals. Acceptance limits should be justified, taking into consideration the pharmacopoeia requirements.
The content uniformity of the premetered DPI dose units should be controlled by a separate test. Typical acceptance criteria are prescribed in pharmacopoeia [9], such as a uniformity-of-dosage-units assay.
9.2.10 Mean Delivered Dose
In inhalation, the dose is a complex concept and attention has to be paid to the precise labeled dose. For pMDIs, DPIs, and MDNs, the content per actuation can be expressed either as metered dose—the quantity of drug contained in the device metering chamber—or as delivered dose—the quantity of drug available to the user, ex device. In the EU, all products containing new chemical entities or containing known drug substances used in inhalation products for the first time should be labeled with the delivered dose or an appropriate alternative (e.g. fine-particle mass). For existing products, current practice in each EU member state should be followed. In any case, it should be clearly stated if the label indication is expressed as metered dose (ex valve), delivered dose (ex actuator), or an appropriate alternative. Different products of the same drug labeled with the same metered or delivered dose might have a different therapeutic effect due to differences in the fine-particle mass.
The amount of drug in one actuation is determined as an average of doses delivered in a dose-uniformity test for a single container. In comparison to a drug-content assay, a wider limit of ±15% of label claim is allowed to illustrate the drug content in one actuation. The mean delivered dose should be expressed as per actuation amount [9].
9.2.11 Number of Actuations Per Container
In metered products with an actuator, the number of actuations for each container should be verified. MDI products can discharge up to several hundred metered doses of drug. Each actuation may contain anywhere from a few micrograms up to milligrams of drug, delivered in a volume typically between 25 and 100 mL. The number of actuations per container should be demonstrated to be not less than the labeled number.
Early fatigue of an actuator or of other related parts of the device, as well as a wrong setting in the locking mechanism on dose-counting, pose the risk of failure to deliver doses from the device during medication. A test can be conducted during testing of the uniformity of delivered dose. Doses should be discharged with intervals not less than 5 seconds between actuations, particularly in pMDIs. Excessively high frequency of actuation may lead to content freezing, and to valve or actuator blockage by frozen matter.
Dose-counting mechanisms are installed in metered-dose inhalers (MDIs) to indicate the remaining dose. In addition to the number of actuations per container, the functionality of the dose counter has to be examined [10].
9.2.12 Fine-Particle Mass
The aerodynamic particle size distribution of the delivered-dose aerosol depends on formulation, device, and patient maneuvers. Determination of the fine-particle mass is requested for all inhalation products and corresponds to the dose of drug assessed in vitro considered suitable for deposition in the lungs. Nebulization products should also be aerodynamically tested if they are formulated as suspensions.
The size of the particles in an inhalation aerosol can express the ability of the drug to reach the intended site of action. The optimum aerodynamic particle size distribution for most inhalation products is generally recognized as being between 1 and 5 μm. The fine-particle mass is then the amount of drug in an inhalation product that has an aerodynamic size considered capable of reaching the lung during inhalation (5 μm and smaller), on a per actuation basis. It is also called the respirable dose of an inhalation product and is often smaller than the delivered dose, due to formulation and administration limitations.
The fine-particle-mass test is conducted using a multistage impactor or impinger apparatus (see Figure 6.1) operating at 28.3 L/min or, in the case of DPIs, at a specific airflow rate dependent on the resistance of the device. The multistage cascade impactor fractionates and collects particles of one or more drug component by aerodynamic diameter through serial multistage impactions. A quantifiable drug mass, from a number of actuations not greater than 10, is discharged into impactor stages corresponding to different aerodynamic sizes of particles through the airflow. Aerodynamically large particles are deposited at the upper stages; aerodynamically small particles will follow the airstream and deposit at lower stages, when an adequate level of momentum is provided to them by discharge acceleration.
During the aerodynamic particle size distribution, the mass balance (total drug substance deposited on surfaces from the valve to the cascade impactor filter) is determined. The total mass of drug collected on all stages and accessories should be between 85 and 115% of label claim on a per actuation basis.
The fine-particle mass is computed as the drug amount pooled from stages corresponding to a particle size distribution less than 5 μm. However, a particle size distribution above 5 μm has to be controlled and analyzed as well, in case this fraction affects the therapeutic index of the inhaled product. Acceptance criteria for particle-size and size-distribution limits are proposed with the aim of characterizing the fine-particle mass of the dose.
The particle size and size distribution of discharged aerosol are described by the particle’s mass median aerodynamic diameter (MMAD) and geometric standard deviation (GSD), calculated from a log-probability plot of the cumulative drug mass fraction against the cut-off diameter of various stages. In all cases, the proposed limits should be qualified by the fine-particle mass results for the batches used in in vivo studies (pivotal, clinical, and/or comparative) and should be reported on a per actuation or per dose basis. A total of five inhalers are used in fine-particle-mass tests, and only the initial discharge is quantified [11].
9.2.13 Spray Pattern and Plume Geometry
A test of spray pattern and plume geometry is conducted during the pharmaceutical development phase of a spray inhalation product. Unless otherwise needed, the plume-geometry test can be excluded as routine for finished-product characterization. “Plume geometry” refers to the shape and size of the spray cloud, whereas “spray pattern” is the size and shape of the spray on a paper sheet.
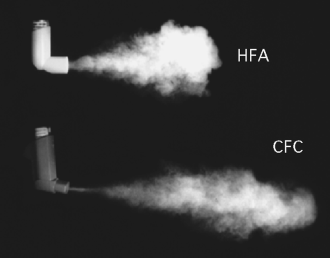
The features of the spray pattern and plume geometry of a metered inhalation product indicate the performance of the valve and/or actuator. A deviation in spray pattern and plume geometry suggests that the uniformity of delivered doses is low and the ability of doses to reach the intended site in the lungs is poor, due to differences in the aerosol shot. Of interest is the difference in plume geometry from that using the same valve in CFC and HFA propellants. The Figure 9.3 shows that the plume of an HFA propellant, having lower pressure, is less elongated, indicating a slower plume motion [3].
Figure 9.3 Plume geometry of HFA and CFC propellants. Courtesy of Dr Andrea Chiesi, Chiesi Farmaceutici, Parma, Italy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


