Purine and Urate Metabolism
Abnormalities of purine metabolism are often found in clinical practice, notably hyperuricaemia and gout. After exploring purine metabolic pathways, this chapter discusses the various disorders of purine metabolism, including their clinical features, diagnosis and treatment.
PURINES
Normal purine metabolism
Urate is the end product of purine metabolism in humans. In most other mammals, it is further metabolized to the more water-soluble allantoin. Humans lack uricase and thus their uric acid levels are higher. It is because of the poor solubility of urate that humans are prone to the clinical effects of hyperuricaemia, such as gout and renal damage. The purines adenine and guanine are constituents of nucleic acid from deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). The purines used by the body for nucleic acid synthesis may be derived from the breakdown of ingested nucleic acid, mostly from cell-rich meat, or they may be synthesized de novo from small molecules. About two-thirds of the body’s urate (3-4 mmol/day) is produced endogenously, with one-third coming from exogenous dietary purines (1-2 mmol/day).
Synthesis of purines
The upper part of Figure 20.1 summarizes some of the more important synthetic steps.
The first step in purine synthesis is condensation of pyrophosphate with phosphoribose to form phosphoribose diphosphate (phosphoribosyl pyrophosphate, PRPP).
The amino group of glutamine is incorporated into the ribose phosphate molecule and pyrophosphate is released. Amidophosphoribosyl transferase catalyses this rate-limiting or controlling step. The enzyme is subject to feedback inhibition by increasing concentrations of purine nucleotides; thus the rate of synthesis is slowed when its products increase. The control of this ratelimiting step may be impaired in primary gout.
Glycine is added to phosphoribosylamine. The rate of purine synthesis is increased in primary gout. In Figure 20.1, the atoms in the glycine molecule have been numbered to correspond with those in the purine and urate molecules, and the solid lines further indicate the final position of the amino acid in these molecules.
Via a series of metabolic steps, purine ribonucleotides (purine ribose phosphates) are formed, which control the second step in the synthetic pathway – the formation of phosphoribosylamine. Ribose phosphate is cleaved off, thereby releasing the purines. Some cytotoxic drugs inhibit various stages of the pathway, thus preventing DNA formation and cell growth.
Fate of purines
Purines synthesized in the body, those derived from the diet and those liberated by endogenous catabolism of nucleic acids may be oxidized to urate or reused for nucleic acid synthesis.
Purines oxidized to urate Some adenine is oxidized to hypoxanthine, which is further oxidized to xanthine. Guanine can also form xanthine. Xanthine, in turn, is oxidized to form urate. The oxidation of both hypoxanthine and xanthine is catalysed by xanthine oxidase in the liver. Thus the formation of urate from purines depends on xanthine oxidase activity: gout may be treated using an inhibitor of this enzyme (allopurinol).
Excretion of urate
Urate is filtered through the glomeruli and most is reabsorbed in the proximal tubules. More than 80 per cent of that formed in urine is derived from more
distal tubular secretion. Urinary excretion is slightly lower in males than in females, which may contribute to the higher incidence of hyperuricaemia in men.
distal tubular secretion. Urinary excretion is slightly lower in males than in females, which may contribute to the higher incidence of hyperuricaemia in men.
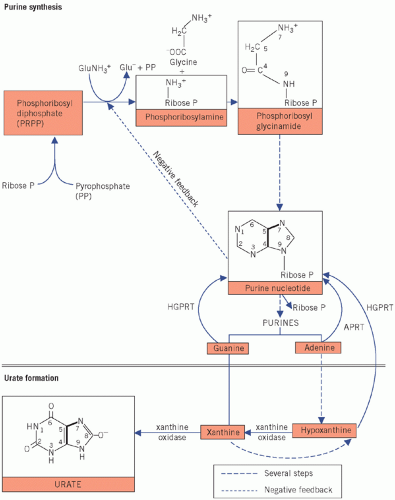 |
Renal secretion may be enhanced by uricosuric drugs (e.g. probenecid or sulfinpyrazone), which block tubular urate reabsorption. Tubular secretion of urates is inhibited by organic acids, such as lactic and oxoacids, and by ketones and thiazide diuretics. Seventy-five per cent of urate leaving the body is in urine. The remaining 25 per cent passes into the intestinal lumen, where it is broken down by intestinal bacteria, the process being known as uricolysis.
HYPERURICAEMIA
Causes of hyperuricaemia
Generally speaking, hyperuricaemia can occur by two main mechanisms: increased production (overproducers) and/or decreased excretion (underexcretors).
The factors that may contribute to hyperuricaemia (summarized in Fig. 20.2) are:
increased synthesis of purines (step 1),
increased intake of purines (step 2),
increased turnover of nucleic acids (step 3),
increased rate of urate formation (step 4),
reduced rate of excretion (step 5).
Increased synthesis, due to impaired feedback control, is probably the most important mechanism causing primary hyperuricaemia, whereas abnormalities of the other steps are causes of secondary hyperuricaemia.
Consequences of hyperuricaemia
Urate is poorly soluble in plasma. At plasma pH, most urate is ionized at position 8 of the purine ring (see Fig. 20.1). This anionic group is associated with the predominant extracellular cation, sodium. Ionization of uric acid decreases as the pH falls, and it therefore becomes less soluble; at a urinary pH below about 6, uric acid may form renal calculi.
Crystallization in joints, especially those of the feet, produces the classic picture of gout. Urate precipitation at these sites causes an inflammatory response with leucocytic infiltration, and it is thought that lactic acid production by these cells causes a local fall in pH: this converts urate to uric acid, which is less soluble than urate, and a vicious circle is set up in which further precipitation, and therefore further inflammation, occurs. In acute attacks of gouty arthritis, local factors are more important than the plasma urate concentration, which is usually normal during the attack.
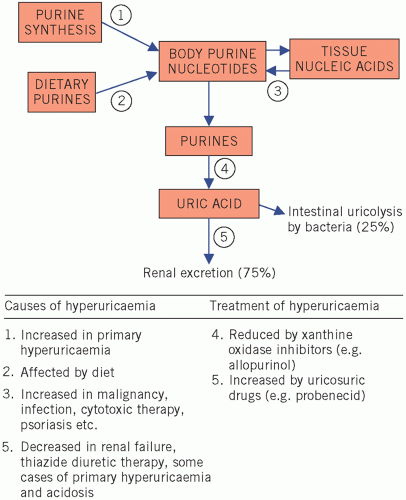 Figure 20.2 Urate metabolism in normal individuals. |
Precipitation may occur in subcutaneous tissues, especially of the ears, and in the olecranon and patellar bursae and tendons. Such deposits are called gouty tophi (Fig. 20.3). A potentially serious effect of hyperuricaemia is precipitation of urate in the kidneys and renal calculi, causing progressive renal damage. For this reason it has been recommended that even asymptomatic cases should probably be treated if the plasma urate concentration is consistently higher than 600 µmol/L. Hyperuricaemia can be primary or secondary.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


