Inborn Errors of Metabolism
The inherited characteristics of an individual are determined by about 50 000 gene pairs, arranged on 23 pairs of chromosomes, one of each pair coming from the father and one from the mother. Genotype diversity is introduced by random selection and recombination during meiosis, as well as by occasional mutation. These genetic variants may at one extreme be incompatible with life, or at the other produce biochemical differences detectable only by special techniques, if at all. Between the two extremes there are many variations that produce functional abnormalities or inborn errors of metabolism (IEM). Incidences of IEM range from about 1 in 100 to 1 in 200 000, depending on the disorder and the population involved.
SOME METABOLIC CONSEQUENCES OF GENETIC DEFECTS
Inherited inborn disorders may involve any peptide or protein, and are usually most obvious if there is an enzyme abnormality. Deficiency of a single enzyme in a metabolic pathway may produce its effects in several ways.
Suppose that substance A is acted on by enzyme X to produce substance B, and that substance C is on an alternative pathway (Fig. 27.1). A deficiency of X may cause:
deficiency of the product (B) of the enzyme reaction, for example cortisol deficiency in congenital adrenal hyperplasia,
accumulation of the substance (A) acted on by the enzyme, for example phenylalanine in phenylketonuria (PKU),
diversion through an alternative pathway: some product(s) of the latter (C) may accumulate and produce effects, for example congenital adrenal hyperplasia when accumulation of androgens causes virilization.
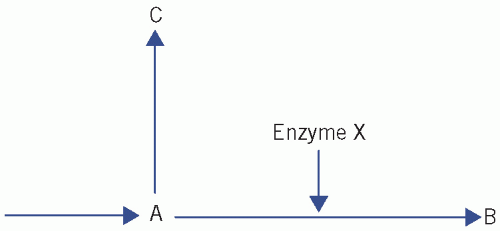 Figure 27.1 Metabolic consequences of genetic defects. |
The effects of the last two types of abnormality will be aggravated if the whole metabolic pathway is controlled by negative feedback from the final product. For example, in congenital adrenal hyperplasia, cortisol deficiency reduces negative feedback, thereby increasing the rate of steroid synthesis and therefore the accumulation of androgens, causing virilization in the female (see Chapter 8).
The clinical effects of some IEM may be modified by, or depend entirely on, physiological or environmental factors. For example, iron loss occurs during menstruation and pregnancy; women with hereditary haemochromatosis accumulate iron less rapidly than men with the same condition and they rarely present with clinical features before the menopause (see Chapter 21). Patients with cholinesterase variants develop symptoms only if the muscle relaxant suxamethonium is given (see Chapter 18).
CLINICAL IMPORTANCE OF INBORN ERRORS OF METABOLISM
Some inborn errors are probably harmless. However, they are important because they produce effects that may lead to misdiagnosis, for example renal glycosuria and Gilbert’s syndrome. In others, it is important to make a diagnosis, even though no effective treatment is yet available.
There is a group of diseases for which recognition in early infancy is of great importance because treatment may prevent irreversible clinical consequences or death. Some of the more important of these are PKU, galactosaemia and maple syrup urine disease.
NEONATAL SCREENING
Many countries have instituted programmes for screening all newborn infants for certain inherited metabolic disorders or congenital defects. The criteria should depend on the following characteristics of the disorder or of the test:
The disease should not be clinically apparent at the time of screening and should have a relatively high incidence in the population screened.
The disease should be treatable or early treatment should improve outcome.
It must be possible to obtain the result of the screening test before irreversible damage is likely to have occurred.
The screening test should be simple and reliable and the cost of the programme should, ideally, be at least partly offset by the cost savings resulting from early treatment. For example, such treatment may sometimes eliminate the need for prolonged institutional care.
Not all these criteria are necessarily fulfilled in all screening programmes.
In the UK, at between 5 and 8 days, babies are screened for certain conditions by taking a small capillary blood sample from a heel prick (see Chapter 26, Fig. 26.2). Blood spots are placed on a paper card, which can be posted to the regional laboratory for assay.
In the UK, screening is generally carried out for neonatal hypothyroidism (see Chapters 11 and 26) and PKU. Other conditions that may be screened for in certain regions include cystic fibrosis, sickle cell disease or thalassaemia, medium-chain acyl coenzyme A dehydrogenase deficiency (MCADD), glucose-6- phosphatase deficiency, galactosaemia and congenital adrenal hyperplasia. (These conditions are discussed elsewhere in this book.) The use of deoxyribonucleic acid (DNA) technology and tandem mass spectroscopy in antenatal screening can be expected to increase in the future.
PRENATAL SCREENING
Prenatal screening, of high-risk groups only, may be performed for some disorders in order to plan the appropriate place and method of delivery for the well-being of the infant or to offer termination, if the diagnosis is made early enough and if it is acceptable.
Prenatal screening for inherited metabolic disorders most commonly involves demonstrating the metabolic defect in cultured fetal fibroblasts obtained by amniocentesis early in the second trimester, or by chorionic villus sampling during the first trimester. Examples of those groups in whom such screening may be indicated include women with a previously affected infant and ethnic groups thought to have a relatively high incidence of the carrier state, such as of Tay-Sachs disease in Ashkenazi Jews. In these high-risk populations, screening is often performed before conception, enabling genetic advice and prenatal diagnosis to be offered to couples who are carriers.
If, as in cystic fibrosis, the gene defect of the parent of an affected infant is known, there may be a case for selective screening of subsequent pregnancies using molecular biological techniques. Prenatal screening for congenital disorders, for example neural tube defects, or chromosomal abnormalities may also be performed.
WHEN TO SUSPECT AN INBORN ERROR OF METABOLISM
The possibility of an inherited metabolic defect should be considered if there are unusual, unexplained clinical features (Box 27.1) or abnormal laboratory findings in
infancy or early childhood, especially if more than one infant in the family has been affected or there has been a consanguineous marriage.
infancy or early childhood, especially if more than one infant in the family has been affected or there has been a consanguineous marriage.
Box 27.1 Some clinical findings suggestive of an inborn error of metabolism
Early
Hypoglycaemia
Metabolic acidosis
Failure to thrive
Vomiting
Fits or spasticity
Hepatosplenomegaly
Prolonged jaundice
A peculiar smell, or staining, of the nappies
Death of child in family and positive family history
Cataracts or retinitis pigmentosa
Late
Intellectual disabilities
Refractory rickets
Renal calculi
Neuropathy
Short stature
Dysmorphic features
Screening tests should be interpreted with caution and a suspected diagnosis confirmed by more specific techniques in a laboratory specializing in such disorders.
Inborn errors presenting acutely are usually due to an enzyme abnormality. This may be demonstrated indirectly, by detecting a high concentration of the substance normally metabolized by the enzyme, or a low concentration of the product; or directly, by demonstrating a low enzyme activity in the appropriate tissue or blood cells; these assays may only be available at special centres. If possible, all cases should be confirmed in this way.
Examples of indirect screening methods include:
estimation of plasma ammonia concentration to test for disorders of the urea cycle or organic acidurias, in which it accumulates,
chromatography of plasma and urine for amino acids for the detection of disorders of amino acid metabolism,
detection of organic acids in urine in disorders of branched-chain amino acid metabolism and organic acidurias.
If the clinical signs and symptoms are strongly suggestive of a particular disorder, specific measurements, such as those of urinary glycosaminoglycan excretion and white cell enzymes characteristic of mucopolysaccharidoses (MPSs), may be used. The technique of tandem mass spectroscopy is proving useful in the investigation of various IEM.
Genetic tests are being used more frequently, and their use is likely to increase still further. Box 27.2 gives a suggested investigation plan.
PRINCIPLES OF TREATMENT OF INBORN ERRORS OF METABOLISM
Some inborn errors can be treated by:
limiting the dietary intake of precursors in the affected metabolic pathway, such as phenylalanine in PKU or lactose in galactosaemia,
supplying the missing metabolic product, such as cortisol in congenital adrenal hyperplasia,
removing or reducing the accumulated product, such as ammonia in urea cycle disorders.
Experimental treatments may be tried for some disorders with particularly poor prognoses. One of these is enzyme replacement by bone marrow transplantation, but the results have sometimes been disappointing and it is not without its own complications. Insertion of the missing or defective gene is being attempted for disorders such as adenosine deaminase deficiency. However, for many disorders there is, at the time of writing, still no treatment unless they respond to one of the measures listed above.
Box 27.2 Possible laboratory investigation of a suspected inborn error of metabolisma
Full blood count
Serum electrolytes, bicarbonate and blood gases for acid-base status
Renal function tests, including plasma urea and creatinine
Liver function tests
Plasma ammonia
Blood glucose
Urine ketones
Serum cholesterol and triglyceride
Plasma lactate
Plasma uric acid
Thyroid function tests
Porphyrins
Further specialist tests
Plasma and sometimes urine amino acids
Urine orotic acid
Urine organic acids
Plasma carnitine
Metabolites in urine or plasma by tandem mass spectroscopy
Specific enzyme assays
DNA analysis of leucocytes or fibroblasts
Histological studies of affected tissue
DISEASES DUE TO INBORN ERRORS OF METABOLISM
Only a few of the known IEM are discussed here. Some of these conditions are mentioned briefly in the relevant chapters in this book.
For the sake of convenience, nine general categories of IEM are arbitrarily defined:
urea cycle defects,
disorders of amino acid metabolism, for example amino acidurias,
lysosomal storage defects,
disorders of carbohydrate metabolism, for example glycogen storage disorders, gluconeogenesis and carbohydrate intolerance defects,
lipid, fatty acid oxidation defects and organic acidurias,
mitochondrial disorders,
peroxisomal disorders,
abnormalities of drug metabolism,
miscellaneous causes.
1 Urea cycle disorders
Urea cycle defects are an important cause of hyperammonaemia and there may also be raised urinary orotic acid concentration, an intermediate metabolite of pyrimidine synthesis derived from carbamyl phosphate. The urea cycle defects can present not only with severe hyperammonaemia, but also with a respiratory alkalosis and low plasma urea concentration (Fig. 27.2). Carbamyl phosphate synthetase (CPS) deficiency is a urea cycle disorder in which, unlike other defects in this pathway, urinary orotic acid is not raised. Ornithine transcarbamylase deficiency is probably the most common urea cycle defect and is sex linked.
CASE 1
A 3-month-old boy was seen in the paediatric out-patient department because of failure to thrive and hypotonia. The family had previously lost a male child, who had died at the age of 9 months. Some of the presenting child’s abnormal biochemistry results were as follows:
Plasma
Sodium 142 mmol/L (135-145)
Potassium 3.8 mmol/L (3.5-5.0)
Urea 0.5 mmol/L (2.5-7.5)
Creatinine 44 µmol/L (40-80)
Ammonia 654 µmol/L (< 20)
Plasma amino acid analysis revealed elevated alanine, glutamine and orotic acid concentrations.
DISCUSSION
The diagnosis was established as ornithine transcarbamylase (OTC) deficiency, one of the urea cycle defects, which is X-linked. Note that the child shows hyperammonaemia and low plasma urea concentration and that he is male, in keeping with a sex-linked condition. There are relatively few causes of a low plasma urea concentration in neonates, and this combined with severe hyperammonaemia supports a diagnosis of a urea cycle disorder. An inborn error of metabolism should be suspected on the death of a baby or when a child presents with failure to thrive, particularly if the parents are consanguineous, for example cousins.
2 Disorders of amino acid metabolism
Many disorders of amino acid metabolism are characterized by raised plasma concentrations of one or more amino acids, with overflow amino aciduria.
The main metabolic pathway for aromatic amino acids is outlined in Figure 27.3, which also indicates the known enzyme defects. Tyrosine, normally produced from phenylalanine, is the precursor of several important substances, inherited disorders of which are considered briefly.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


