Pulmonary Arterial Hypertension
KEY CONCEPTS
![]() Pulmonary arterial hypertension (PAH) is defined as a mean pulmonary artery pressure (mPAP) ≥25 mm Hg at rest with a pulmonary wedge pressure (also known as pulmonary artery occlusion pressure) or left ventricular end-diastolic pressure (LVEDP) ≤15 mm Hg measured by right cardiac catheterization.
Pulmonary arterial hypertension (PAH) is defined as a mean pulmonary artery pressure (mPAP) ≥25 mm Hg at rest with a pulmonary wedge pressure (also known as pulmonary artery occlusion pressure) or left ventricular end-diastolic pressure (LVEDP) ≤15 mm Hg measured by right cardiac catheterization.
![]() Diagnosis of PAH is growing due to increased awareness and knowledge of the disease state, leading to earlier and improved evaluation and identification.
Diagnosis of PAH is growing due to increased awareness and knowledge of the disease state, leading to earlier and improved evaluation and identification.
![]() Regardless of the etiology, be it unknown or related to an associated medical condition, subgroups of PAH are based on similar clinical and pathologic physiology.
Regardless of the etiology, be it unknown or related to an associated medical condition, subgroups of PAH are based on similar clinical and pathologic physiology.
![]() The underlying cause of PAH is a complicated amalgam of endothelial cell dysfunction, a procoagulant state, platelet activation, vasoconstriction, loss of relaxing factors, cellular proliferation, hypertrophy, fibrosis, and inflammation.
The underlying cause of PAH is a complicated amalgam of endothelial cell dysfunction, a procoagulant state, platelet activation, vasoconstriction, loss of relaxing factors, cellular proliferation, hypertrophy, fibrosis, and inflammation.
![]() Patients with PAH present with exertional dyspnea, fatigue, weakness, and exertion intolerance. As the disease progresses, symptoms of right heart dysfunction and failure, such as dyspnea at rest, lower extremity edema, chest pain, and syncope, are seen.
Patients with PAH present with exertional dyspnea, fatigue, weakness, and exertion intolerance. As the disease progresses, symptoms of right heart dysfunction and failure, such as dyspnea at rest, lower extremity edema, chest pain, and syncope, are seen.
![]() The only way to make a definitive diagnosis of PAH is by right heart catheterization. The right heart catheterization provides important prognostic information and can be used to assess pulmonary vasoreactivity prior to initiating therapy.
The only way to make a definitive diagnosis of PAH is by right heart catheterization. The right heart catheterization provides important prognostic information and can be used to assess pulmonary vasoreactivity prior to initiating therapy.
![]() The goals of treatment are to alleviate symptoms, improve the quality of life, slow the progression of the disease, and improve survival.
The goals of treatment are to alleviate symptoms, improve the quality of life, slow the progression of the disease, and improve survival.
![]() A general goal of PAH treatment is to correct the imbalance between vasoconstriction and vasodilation and prevent adverse thrombotic events to improve oxygenation and quality of life.
A general goal of PAH treatment is to correct the imbalance between vasoconstriction and vasodilation and prevent adverse thrombotic events to improve oxygenation and quality of life.
![]() Nonpharmacologic therapy is frequently used to address comorbid conditions that often accompany PAH.
Nonpharmacologic therapy is frequently used to address comorbid conditions that often accompany PAH.
![]() Conventional therapy of PAH includes oral anticoagulants, diuretics, oxygen, and digoxin.
Conventional therapy of PAH includes oral anticoagulants, diuretics, oxygen, and digoxin.
![]() Prostacyclin analogs such as epoprostenol, treprostinil, and iloprost induce potent vasodilation of pulmonary vascular beds.
Prostacyclin analogs such as epoprostenol, treprostinil, and iloprost induce potent vasodilation of pulmonary vascular beds.
![]() Endothelin receptor antagonists, bosentan and ambrisentan, improve exercise capacity, hemodynamics, and functional class in PAH.
Endothelin receptor antagonists, bosentan and ambrisentan, improve exercise capacity, hemodynamics, and functional class in PAH.
![]() Phosphodiesterase-5 inhibitors, including sildenafil and tadalafil, are potent and highly specific drugs that have been shown to reduce mPAP and improve functional class.
Phosphodiesterase-5 inhibitors, including sildenafil and tadalafil, are potent and highly specific drugs that have been shown to reduce mPAP and improve functional class.
![]() Combination therapy in PAH may address more than one mechanism causing this disease. Combination therapy in clinical trials has provided additional benefit, but more studies are needed.
Combination therapy in PAH may address more than one mechanism causing this disease. Combination therapy in clinical trials has provided additional benefit, but more studies are needed.
Pulmonary hypertension is a term describing a group of conditions relating to elevated blood pressure measured within the pulmonary artery. Pulmonary hypertension is not a specific diagnosis; rather it is a complex group of disorders relating to the pulmonary circulation. Pulmonary hypertension is classified into five groups according to the World Health Organization (WHO; see Table 17-1).1 Pulmonary arterial hypertension (PAH) or Group 1 pulmonary hypertension is a progressive disease characterized by an elevation in pulmonary arterial pressure and pulmonary vascular resistance. ![]() PAH may be defined as a mean pulmonary artery pressure (mPAP) ≥25 mm Hg at rest, with a pulmonary wedge pressure (also known as a pulmonary artery occlusion pressure or left ventricular end-diastolic pressure [LVEDP]) ≤15 mm Hg measured by cardiac catheterization.2
PAH may be defined as a mean pulmonary artery pressure (mPAP) ≥25 mm Hg at rest, with a pulmonary wedge pressure (also known as a pulmonary artery occlusion pressure or left ventricular end-diastolic pressure [LVEDP]) ≤15 mm Hg measured by cardiac catheterization.2
TABLE 17-1 World Health Organization Classification of Pulmonary Hypertension
PAH may occur in the setting of underlying medical conditions or as an idiopathic disease (idiopathic PAH [IPAH]). Historically, medical treatment of PAH has been limited due to lack of effective, targeted therapy. Without medical therapy, IPAH portends a poor prognosis (median survival 2.8 years) after diagnosis.3 Prior to the availability of disease-specific therapy for IPAH, survival rates for 1, 3, and 5 years were 68%, 48%, and 34%, respectively.4 Since the approval of epoprostenol in 1995, a number of new therapeutic options have been developed. In a recent epidemiologic study, survival rates for 1, 2, and 3 years in patients on targeted therapy were 83%, 67%, and 58%, respectively.5
EPIDEMIOLOGY
The prevalence of PAH is estimated to be 15 to 50 patients per million individuals. Unfortunately, only 15,000 to 20,000 of the afflicted patients have an established diagnosis of PAH and are currently receiving treatment. In a French registry study of more than 600 patient with PAH, Humbert found that the most common cause of PAH was IPAH (approximately 40%), followed by PAH associated with connective tissue diseases (15.3%), congenital heart disease (11.3%), portal hypertension (10.4%), and familial PAH (FPAH) (3.9%).6 ![]() However, diagnosis of PAH is growing due to increased awareness and knowledge of the disease state, leading to earlier and improved evaluation and identification.
However, diagnosis of PAH is growing due to increased awareness and knowledge of the disease state, leading to earlier and improved evaluation and identification.
ETIOLOGY
PAH most often originates with a predisposing state and one or more inciting factors that could be genetic or environmental exposures.7 Once a permissive environment exists, multiple mechanisms can be activated leading to vascular constriction, cellular proliferation, and a prothrombotic state resulting in PAH and its sequelae.8 PAH can be associated with numerous conditions as well as being an idiopathic condition (IPAH). The incidence of IPAH is estimated to be 5 to 6 per 1 million in North America and Europe, with a marked female predominance (male-to-female ratio, 1:1.7), and mean age at time of recognition is approximately 37 years, although there is considerable variation.8,9 Although uncommon in the United States, the commonest form of PAH worldwide is schistosomiasis followed by congenital heart disease and pulmonary hypertension of early childhood.10 Rheumatologic diseases such as scleroderma, systemic lupus erythematosus, rheumatoid arthritis, and myositis are also associated with development of PAH. Patients with scleroderma who develop PAH, estimated between 7% and 12% of patients, have markedly worse outcomes in comparison to other PAH subgroups. Patients with human immunodeficiency virus (HIV) infection can develop PAH with a prevalence of 0.5%. In patients with liver disease, portal hypertension may cause concurrent pulmonary hypertension in an estimated 2% to 6% of patients.11 Multiple drugs and toxins have been associated with PAH but those that definitively precipitate PAH include anorexigens such as aminorex, fenfluramine, and dexfenfluramine.10,12 Other drugs considered to be likely or possible causative agents for PAH include amphetamines, L-tryptophan, cocaine, and certain chemotherapeutic agents (mitomycin C, carmustine, etoposide, cyclophosphamide, bleomycin).9 Heritable PAH (HPAH) includes both IPAH with germline mutations and familial cases without an identified mutation. Germline mutations seen in PAH include bone morphogenetic protein receptor 2 (BMPR2) and activin receptor-like kinase 1 (ALK-1). Genetic testing for these mutations may be offered and professional genetic counseling should be provided.11
PATHOPHYSIOLOGY
![]() Regardless of etiology, all subgroups of PAH are based on similar clinical and pathologic physiology.
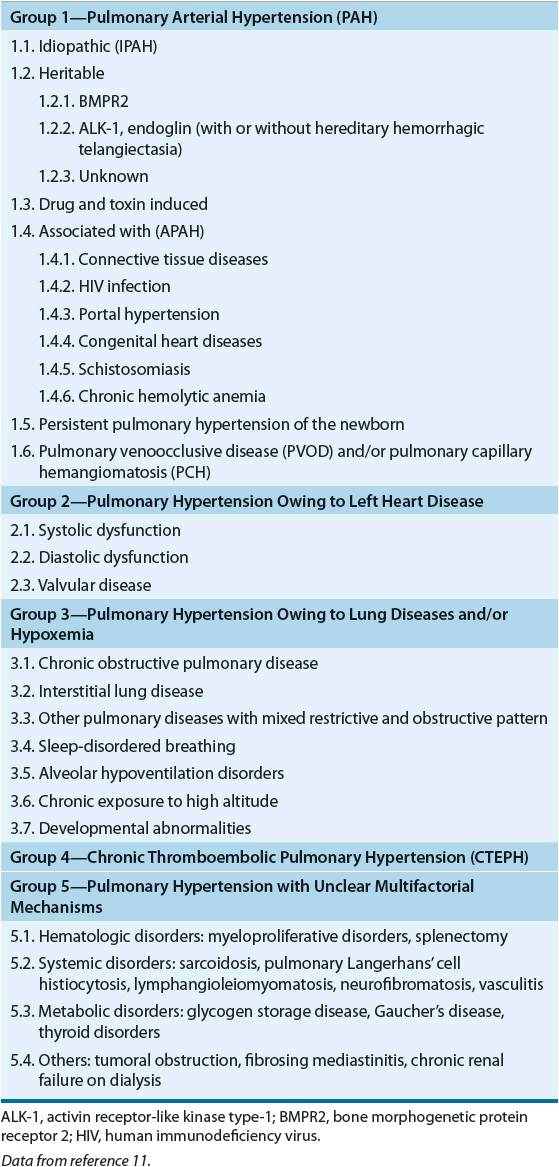
Regardless of etiology, all subgroups of PAH are based on similar clinical and pathologic physiology. ![]() The pathobiology of PAH involves several key biologic events, including endothelial cell dysfunction, a procoagulant state, platelet activation, constricting factors, loss of relaxing factors, cellular proliferation, hypertrophy, fibrosis, and inflammation—all combining to produce progressive and deleterious vascular remodeling (Fig. 17-1).13,14 Multiple genetic mutations are known to contribute to the pathophysiology of PAH, including BMPR2, ALK-1, nitric oxide synthase (ec-NOS), carbamoyl-phosphate synthase gene, and 5-hydroxytryptamine (serotonin [5-HT]) transporter (5-HTT).13,15 A mutation of BMPR2 receptor is an aberration of signal transduction in the pulmonary vascular smooth muscle cell that is postulated to alter apoptosis favoring cellular proliferation. ALK-1 is part of the transforming growth factor-β superfamily and is seen in hereditary hemorrhagic telangiectasia and PAH.16 5-HTT is associated with pulmonary artery smooth muscle proliferation and is present in IPAH in the homozygous form in 65% of patients.17 Dysregulation of 5-HT synthesis mediated via tryptophan hydroxylases is closely linked to the hypoxic PAH phenotype in mice and may contribute to PAH development.18
The pathobiology of PAH involves several key biologic events, including endothelial cell dysfunction, a procoagulant state, platelet activation, constricting factors, loss of relaxing factors, cellular proliferation, hypertrophy, fibrosis, and inflammation—all combining to produce progressive and deleterious vascular remodeling (Fig. 17-1).13,14 Multiple genetic mutations are known to contribute to the pathophysiology of PAH, including BMPR2, ALK-1, nitric oxide synthase (ec-NOS), carbamoyl-phosphate synthase gene, and 5-hydroxytryptamine (serotonin [5-HT]) transporter (5-HTT).13,15 A mutation of BMPR2 receptor is an aberration of signal transduction in the pulmonary vascular smooth muscle cell that is postulated to alter apoptosis favoring cellular proliferation. ALK-1 is part of the transforming growth factor-β superfamily and is seen in hereditary hemorrhagic telangiectasia and PAH.16 5-HTT is associated with pulmonary artery smooth muscle proliferation and is present in IPAH in the homozygous form in 65% of patients.17 Dysregulation of 5-HT synthesis mediated via tryptophan hydroxylases is closely linked to the hypoxic PAH phenotype in mice and may contribute to PAH development.18
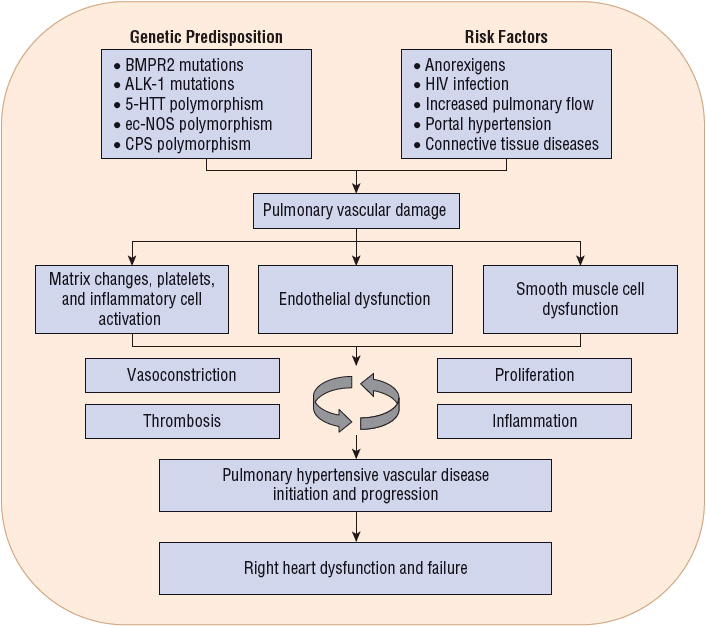
FIGURE 17-1 Pulmonary arterial hypertension; potential pathogenetic and pathobiologic mechanisms. (ALK-1, activin receptor-like kinase 1 gene; BMPR2, bone morphogenetic protein receptor 2 gene; CPS, carbamoyl-phosphate synthase gene; ec-NOS, nitric oxide synthase gene; 5-HTT, serotonin transporter gene; HIV, human immunodeficiency virus. (Reproduced with permission from reference 30.)
Molecular, cellular, and genetic mechanisms are mediated by a variety of biologically active compounds, including prostacyclin (PGI2), endothelin-1 (ET-1), nitric oxide (NO), and 5-HT. PGI2 is a vasodilatory and antiproliferative substance that is produced by the endothelial cells, and the synthesis of PGI2 and its circulating levels are reduced in PAH. Furthermore, thromboxane, a vasoconstrictor, is increased in PAH. ET-1 is produced in the endothelium, and it possesses potent vasoconstrictor and mitogenic effects. ET-1 levels are increased in PAH and clearance is reduced. ET-1 acts via the endothelin receptors (ETA and ETB) to promote vascular smooth muscle proliferation and vasoconstriction.14,19 Plasma levels of ET-1 are correlated with severity of PAH and prognosis.20 NO is produced in the endothelium via NO synthase and leads to vasodilation and opening of cell membrane potassium channels to allow potassium ion efflux, membrane depolarization, and calcium channel inhibition. Voltage-dependent potassium channels are inhibited by a number of stimuli that promote PAH, including hypoxia and fenfluramine, resulting in downregulated potassium channels in patients with PAH. Entering calcium is a signal for release of sarcoplasmic calcium and activation of the contractile apparatus. NO promotes vasodilation through calcium channel inhibition. In PAH there is evidence of decreased NO synthase expression, leading to vasoconstriction and cell proliferation.21 Elevated 5-HT has been observed and vasoconstriction mediated via the increased expression of the 5-HT1B receptor is seen in PAH.8
Autoantibodies, proinflammatory cytokines, and inflammatory infiltrates may also participate in the pathogenesis of PAH. Coagulation is disordered in PAH as evidenced by increased levels of von Willebrand factor, plasma fibrinopeptide A, plasminogen activator inhibitor-1, 5-HT, and thromboxane. Furthermore, tissue plasminogen activator, thrombomodulin, NO, and PGI2 are decreased, leading to an imbalance favoring thrombosis. Endothelial dysfunction is the common denominator of mechanisms for PAH, and a variety of injuries, such as shear stress, inflammation, toxins, and hypoxia, are thought to be involved.10,13
![]() The signs and symptoms of PAH are highly variable depending on the stage of the disease and comorbidities (Table 17-2). Symptoms may include exertional dyspnea, fatigue, and weakness. As the disease progresses, patients may experience dyspnea at rest, chest pain, presyncope, syncope, lower extremity edema, and abdominal bloating and distension. On physical exam, patients with PAH may have an accentuated component of S2 audible at the apex of the heart, midsystolic ejection murmur, palpable left parasternal lift, right ventricular S4 gallop, and a prominent “a” wave.10 Hepatojugular reflux, a diastolic murmur of pulmonary regurgitation, and a systolic murmur of tricuspid regurgitation may be present in advanced disease.9
The signs and symptoms of PAH are highly variable depending on the stage of the disease and comorbidities (Table 17-2). Symptoms may include exertional dyspnea, fatigue, and weakness. As the disease progresses, patients may experience dyspnea at rest, chest pain, presyncope, syncope, lower extremity edema, and abdominal bloating and distension. On physical exam, patients with PAH may have an accentuated component of S2 audible at the apex of the heart, midsystolic ejection murmur, palpable left parasternal lift, right ventricular S4 gallop, and a prominent “a” wave.10 Hepatojugular reflux, a diastolic murmur of pulmonary regurgitation, and a systolic murmur of tricuspid regurgitation may be present in advanced disease.9
TABLE 17-2 World Health Organization Functional Classification of Pulmonary Arterial Hypertension (PAH)

Several comorbidities and environmental factors play a role in the development of PAH and must be evaluated when establishing an initial diagnosis of PAH (Fig. 17-2). In patients with a clinical suspicion of PAH, Doppler echocardiography should be performed as a noninvasive screening test that can detect increased pulmonary pressures, although this study cannot be used to definitively diagnose PAH.22 Echocardiography can also be used to assess treatment interventions and to follow disease progression.10 ![]() However, right heart catheterization is the definitive study to use in diagnosis of PAH and when patients are worsening clinically.15 Right heart catheterization provides important prognostic information and can be used to assess pulmonary vasoreactivity with the administration of fast-acting, short-duration vasodilators to determine the extent of vascular smooth muscle constriction and vasodilator response to calcium channel blockers (CCBs; strength of recommendation: A for IPAH; E/C for associated pulmonary arterial hypertension [APAH]).9 Table 17-3 lists the grading criteria for recommendations, and Table 17-4 lists commonly used agents and their dosages. The consensus definition of a positive response is defined as a reduction of mPAP by at least 10 mm Hg to a value of 40 mm Hg or less.23 Patients with an acute response (approximately 13% on initial testing) are most likely to have a beneficial hemodynamic and clinical response. These patients may be able to be treated with CCBs. However, about half of these patients lose an acute vasodilator response when tested 1 year later.24 Therefore, even this small group of patients who may be treated with CCBs must be followed closely for safety and efficacy. If the patient loses the acute vasodilator response, he or she needs to be switched to different PAH therapy. Patients who have a negative response on initial vasodilator testing are not candidates for treatment with CCBs.9,25
However, right heart catheterization is the definitive study to use in diagnosis of PAH and when patients are worsening clinically.15 Right heart catheterization provides important prognostic information and can be used to assess pulmonary vasoreactivity with the administration of fast-acting, short-duration vasodilators to determine the extent of vascular smooth muscle constriction and vasodilator response to calcium channel blockers (CCBs; strength of recommendation: A for IPAH; E/C for associated pulmonary arterial hypertension [APAH]).9 Table 17-3 lists the grading criteria for recommendations, and Table 17-4 lists commonly used agents and their dosages. The consensus definition of a positive response is defined as a reduction of mPAP by at least 10 mm Hg to a value of 40 mm Hg or less.23 Patients with an acute response (approximately 13% on initial testing) are most likely to have a beneficial hemodynamic and clinical response. These patients may be able to be treated with CCBs. However, about half of these patients lose an acute vasodilator response when tested 1 year later.24 Therefore, even this small group of patients who may be treated with CCBs must be followed closely for safety and efficacy. If the patient loses the acute vasodilator response, he or she needs to be switched to different PAH therapy. Patients who have a negative response on initial vasodilator testing are not candidates for treatment with CCBs.9,25
FIGURE 17-2 Evaluation of causes of PAH. (Adapted from Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493–2537.)
TABLE 17-3 Relationship of Strength of Recommendation Scale to Quality of Evidence and Net Benefit

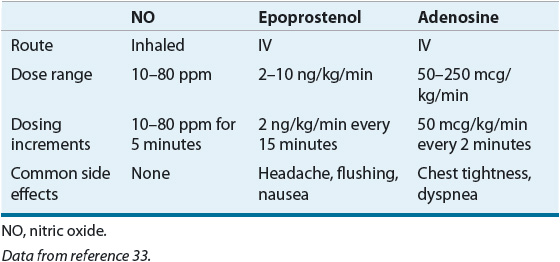
TABLE 17-4 Agents for Vasodilator Testing in Pulmonary Arterial Hypertension
Because PAH commonly occurs in the setting of connective tissue disease, serologic markers should be obtained to confirm or exclude these diagnoses.9,26 Liver function tests (LFTs) should also be evaluated due to the increased risk for PAH in patients with cirrhosis and portal hypertension. HIV is associated with an increased prevalence of PAH, and HIV testing should be done as part of the initial PAH workup.9 Chronic thromboembolic pulmonary hypertension (CTEPH) should be evaluated with ventilation–perfusion lung scans and/or pulmonary angiography. Pulmonary function testing and arterial blood oxygenation should be evaluated. The diffusing capacity of carbon monoxide may be particularly helpful in systemic sclerosis and PAH.10 In patients with PAH, serial determinations of functional class, exercise capacity (assessed by the 6-minute walk distance), and serial biomarkers provide benchmarks for disease severity, response to therapy, and progression.9,26
CLINICAL PRESENTATION Pulmonary Arterial Hypertension
< div class='tao-gold-member'>



