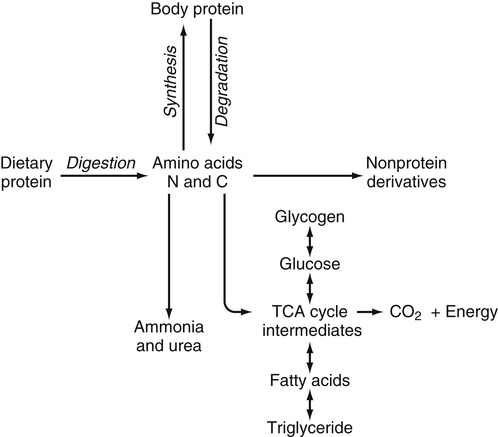
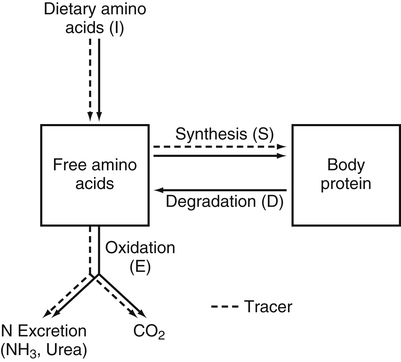
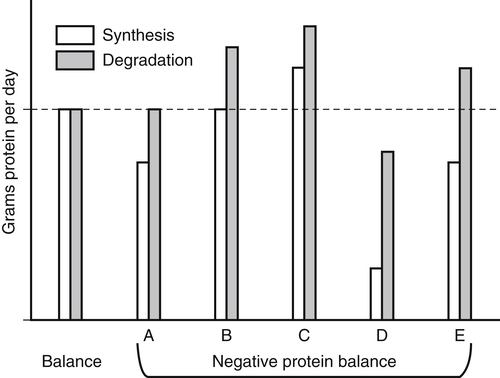
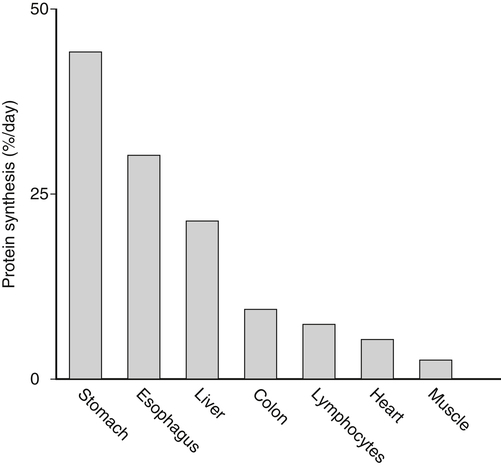
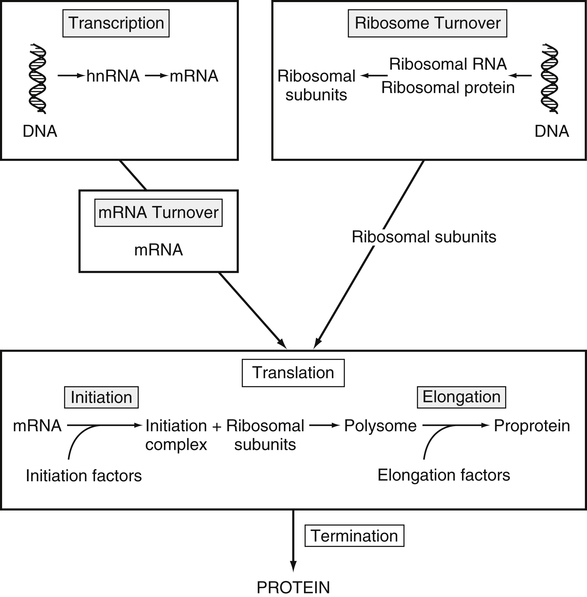
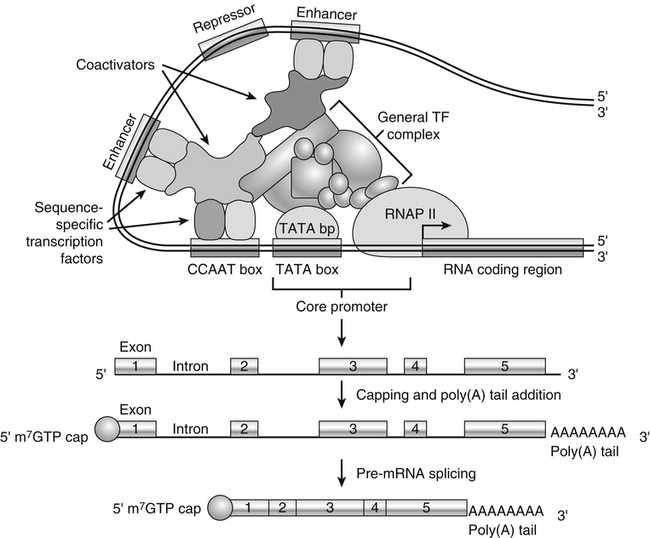
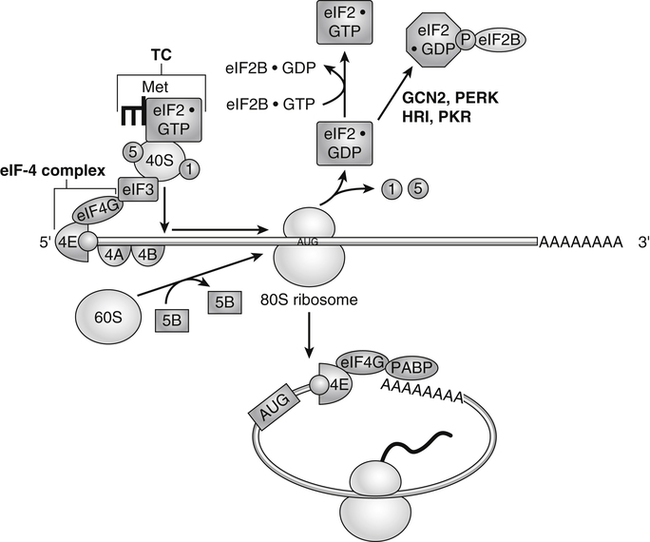
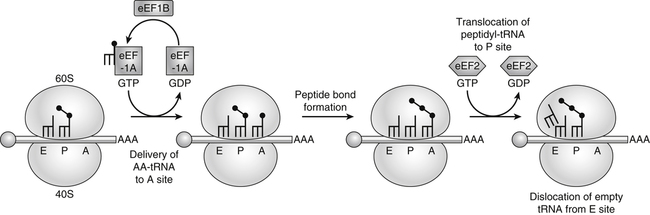
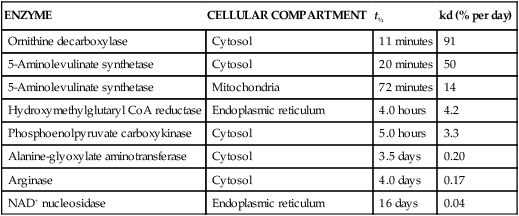
Tracy G. Anthony, PhD and Margaret McNurlan, PhD The interchange between body protein and the pool of free amino acids is depicted schematically in Figure 13-1. The process by which body protein is continually degraded and resynthesized is called protein turnover, a term used collectively to include both protein synthesis and protein degradation. The tripeptide glutathione (γ-glutamylcysteinylglycine) is unusual in that its synthesis accounts for a large amount of the body’s cysteine flux. As for amino acids in proteins, most of the amino acids in glutathione are returned to the amino acid pool upon glutathione turnover. Glutathione synthesis is catalyzed by cytosolic enzymes, not by the ribosomal machinery, and is discussed further in Chapter 14. In addition to serving as substrates for protein synthesis, amino acids also are degraded to provide compounds that can enter central pathways of fuel metabolism, and small amounts are converted to nonprotein compounds. For most adults who are in protein balance, the amount of amino acids degraded is essentially equivalent to the amount in the diet. The degradative pathways are also shown schematically in Figure 13-1. Degradation involves the removal of nitrogen, primarily as urea and ammonia, and the catabolism of the carbon skeleton. The end result of the degradation of the carbon skeleton of amino acids is the provision of energy either directly or through the formation of compounds such as glucose and fatty acids, which can then be stored or metabolized to provide energy. The pathways for the oxidative metabolism of amino acids and nitrogen excretion are discussed in detail in Chapter 14, but it is important to understand the integrated nature of protein metabolism that is represented by Figure 13-1. The needs of the body regulate the flux of amino acids through these possible pathways; that is, whether amino acids are used for the synthesis of protein, oxidized for energy, or used to form glucose. There are also pathways within the body for conversion of amino acids to end products other than protein. These reactions are depicted in Figure 13-1 as nonprotein derivatives. Nonprotein derivatives include compounds such as purine and pyrimidine bases, neurotransmitters such as serotonin, nonpeptide hormones such as catecholamines, and other specialized compounds such as carnitine. Because the amounts of amino acids irreversibly consumed in the synthesis of nonprotein compounds are normally much smaller than those consumed either by protein synthesis or by amino acid oxidation, these pathways often are ignored in the assessment of protein turnover and nitrogen balance. However, the amounts of some of these compounds that are synthesized can be substantial (e.g., creatine, heme, and nucleic acids), and for some amino acids, synthesis of these nonprotein compounds can represent a significant portion of total amino acid utilization during periods of protein deprivation. The pathways shown in Figure 13-1 can be simplified to focus specifically on the interactions of amino acids with body protein through protein synthesis and protein degradation (Figure 13-2). In this simplified scheme, all the tissue and circulating proteins are considered together, and likewise the free amino acid pool is reduced to a single, homogeneous pool, rather than the complex arrangements of pools in blood, individual tissues, and subcellular compartments that are known to exist. This simplification has proved helpful in conceptualizing and developing methods for measuring the exchange of amino acids between the free amino acid pool and the protein pool. This simple model in Figure 13-2 highlights the exchange of free amino acids with body protein through the processes of protein synthesis and protein degradation, and also the entry and exit of amino acids by dietary intake and oxidation. Essential amino acids enter the body free pool from the digestion and absorption of dietary protein (I) and from the degradation of body protein (D). Removal of amino acids from the free pool occurs either by the synthesis of protein (S) or through excretion (E) via oxidation to CO2 with the concurrent excretion of nitrogen, mainly as ammonia and urea. If the amounts of free amino acids in the pool are constant, then the sum of the processes that remove amino acids (protein synthesis and catabolism) is equal to the sum of the processes by which amino acids enter the free pool (from protein degradation and dietary protein intake). Q, the sum of the rates of either entry or exit from the free amino acid pool, has been termed the flux rate. This is sometimes also known as the rate of appearance, Ra, or rate of disappearance, Rd. In an adult in nitrogen equilibrium or protein balance, nitrogen intake (I) is equal to nitrogen excretion (E), and protein synthesis (S) is equal to protein degradation (D). For an individual to be in positive nitrogen balance, there must be net protein synthesis or accretion (S > D), whereas there must be net protein degradation or loss for an individual to be in negative nitrogen balance (S < D). From the aforementioned relationships, it is clear that protein is retained in the body when synthesis exceeds degradation, and that protein is lost from the body when degradation exceeds synthesis. As shown in Figure 13-3, loss of body protein can occur from a decrease in the synthesis of protein with no change in protein degradation (Figure 13-3, A), an increase in the degradation with no change in protein synthesis (Figure 13-3, B), from either an increase (Figure 13-3, C) or a decrease (Figure 13-3, D) in both synthesis and degradation with protein degradation exceeding protein synthesis, or from an increase in degradation along with a decrease in synthesis (Figure 13-3, E). In a number of pathological conditions, body protein degradation exceeds synthesis, with both protein synthesis and degradation rates elevated over the rates in healthy individuals. In the case of infection in malnourished children, body protein is lost, but both synthesis and degradation rates are depressed. In early starvation, net loss of lean body mass is due to an increase in protein degradation along with a decrease in protein synthesis. Likewise, positive protein balance can be achieved by increases in protein synthesis, by decreases in protein degradation, or with changes in both protein synthesis and degradation, such that synthesis exceeds degradation. For example, in children recovering from burn injury, both the rates of protein synthesis and degradation were increased, but the increase in synthesis was larger than the increase in protein degradation (Borsheim et al., 2010; also see Figure 13-12 later in this chapter). Although the illustration of protein turnover in Figure 13-2 is presented in terms of whole-body protein, the balance between the processes of synthesis and degradation also determines the net protein balance at the level of individual tissues or organs and for individual proteins. Examples of this type of regulation are discussed later in this chapter. Protein turnover allows the body to degrade and replace proteins that are oxidized, damaged, misfolded, or otherwise nonfunctional. It also allows the body to change the relative amounts of different proteins to respond to changes in nutritional and physiological conditions. Individual proteins vary in their rate of turnover, as shown in Table 13-1 for several examples of individual liver proteins. Relatively high rates of turnover of regulatory proteins allow for more rapid adaptation in the levels of these proteins in response to changing conditions. Levels of proteins with very slow turnover rates, such as collagen with a half-life of approximately 300 days, remain relatively constant. TABLE 13-1 Turnover Rates of Enzymes in Rat Liver∗ ∗Turnover rates are expressed as half-lives (t½, the time to replace half the molecules originally present) and fractional turnover rates (kd, percent turned over per day). Data from Waterlow, J. C., Garlick, P. J., & Milward, D. J. (1978). Protein turnover in mammalian tissues and in the whole body (pp. 490–492). Amsterdam: North-Holland Publishing. At the level of individual tissues, higher turnover rates are associated with a more rapid cellular turnover rate (e.g., epithelial cells of small intestine) or the capacity of a tissue to respond more rapidly to changes in the environment (e.g., liver). The turnover rates for a selection of human tissues are shown in Figure 13-4. The high rates of protein synthesis in the stomach and esophagus reflect both the secretory function and the rapid replacement of cells of the gastric and esophageal mucosa. The liver also has a relatively high rate of turnover, which facilitates adaptation to changes such as alterations in nutrient intake. By contrast, the rate of protein synthesis is relatively slower in muscle tissue, and changes in protein composition of this tissue in response to altered conditions (e.g., work-induced hypertrophy) occur more slowly. The most inclusive definition of protein synthesis describes the processes required for a gene to be transcribed, processed, translated, folded, and modified and localized, if necessary, to generate a fully functional protein. Each process comprises multiple steps, and regulation can occur at one or more of the steps within each process, as outlined in Figure 13-5. Major advances in understanding the regulation of protein synthesis at the molecular level have been made in the last two decades. Furthermore, with whole genome sequences now available for a growing number of different organisms, including rodents and humans, new information clarifying and extending our current understanding of the regulation of protein synthesis is accumulating at a faster rate than ever before. The revelation that the human genome contains approximately 30,000 genes, only about twice as many as found in invertebrates, has caused the scientific community to reevaluate the influence of the genome in determining animal variety and organismal complexity. It is now believed that the regulation of protein synthesis is the driver in determining cellular diversity. In eukaryotic cells, the ability to express biologically active or functional proteins comes under major regulation at several points: deoxyribonucleic acid (DNA) transcription, ribonucleic acid (RNA) processing, messenger RNA (mRNA) stability, mRNA translation, and posttranslational protein modifications and folding. Because of their complexity, only a basic overview of the processes involved in protein synthesis and its regulation are given in this chapter. 1. Messenger RNAs (mRNAs), which are used by the translational machinery to determine the order of amino acids incorporated into an elongating polypeptide during the process of mRNA translation. 2. Transfer RNAs (tRNAs), which carry individual amino acids to the mRNA template, thereby allowing correct insertion of amino acids into the growing polypeptide chain. 3. Ribosomal RNAs (rRNAs), which are assembled with numerous ribosomal proteins to form the ribosomes, and which, in eukaryotic cells, include (designated by centrifugal sedimentation size) the 28S, 5S, and 5.8S rRNAs that are associated with the large (60S) ribosomal subunit and the 18S rRNA that is associated with the small (40S) ribosomal subunit. 4. Regulatory RNAs such as microRNAs (miRNAs) and endogenous small interfering RNAs (siRNAs), which decrease protein expression by increasing mRNA degradation or reducing mRNA translation. 5. Small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs), which are involved in modifying other RNAs within the nucleus. 6. Other RNAs, including Piwi-interacting RNAs (piRNAs), antisense RNAs, and long noncoding RNAs, all of which can play diverse roles in regulating gene expression. RNA synthesis, as illustrated in Figure 13-6, is catalyzed by a family of enzymes called RNA polymerases (RNAPs). RNAP requires a DNA template to synthesize RNA. Transcription of the different classes of RNAs in eukaryotes is carried out by three different RNAPs. RNAP I synthesizes the rRNAs that comprise the major RNA components of ribosomes. RNAP II synthesizes mRNA precursors and most miRNAs and snRNAs. RNAP III synthesizes the tRNAs, 5S rRNA, and other small RNAs. All RNAPs contain a core group of proteins that make up the basic enzymatic unit required for transcription to proceed. In addition, multisubunit protein cofactors have been identified that associate with the various RNAP core proteins to increase enzyme stability and regulate transcriptional activity. Short nucleotide sequences within the DNA template that regulate the rate of RNA synthesis are called cis-acting elements. Cis-elements that facilitate the initiation of transcription are of two types, termed promoters and enhancers. Promoters are sequences that closely precede the transcription start site and increase the ability of RNAP II to recognize the site at which initiation begins. Almost all eukaryotic genes contain core promoters of two types that are termed CCAAT boxes and TATA boxes, names that are based on their conserved nucleotide sequences (see Figure 13-6). In addition to core promoters, proximal and distal promoter sequences enhance RNAP II activity even further. Enhancer elements are regulatory sequences that activate one or more genes in a cluster. These cis-elements can be located either upstream or downstream of the transcription start site. Other regulatory sequences inhibit or downregulate transcription and are called repressor domains. The number and type of cis-elements vary among genes. Proteins that bind cis-elements are termed trans-acting factors, or, more commonly, transcription factors. Transcription factors are DNA-binding proteins that can enhance or repress gene expression. Recent estimates suggest there are 1,700 to 1,900 transcription factors in humans (Vaquerizas et al., 2009). Transcription factors bind proximal or distal promoter regions, whereas still others interact with enhancer or repressor elements. For example, a family of proteins identified as TF (for general transcription factors regulating RNAP) interact with the TATA box, and the protein identified as C/EBP (for CCAAT/enhancer binding protein) binds to the CCAAT box element. When binding cis-elements, a transcription factor often pairs up with a second DNA-binding protein. When the second DNA-binding protein is identical, a homodimer is formed; when the second DNA-binding protein is different, a heterodimer is formed. The absolute numbers, ratios, and combinations of transcription factors that interact with regulatory sequences on DNA all impact control of RNA synthesis. This along with the presence of multiple cis-acting elements for each template strand of DNA results in a diverse array of binding sites for different regulatory proteins, revealing substantial combinatorial complexity. It also affords a means of coordinately regulating a number of genes. For example, the pathway for the synthesis of cholesterol (see Chapter 17) involves at least 23 enzymes, and many of the genes for enzymes in this pathway are regulated by a family of transcription factors called sterol regulatory element binding proteins (SREBPs). Another family of transcription factors responsible for coordinate regulation of genes are the retinoic acid (RAR) and 9-cis-retinoic acid (RXR) nuclear receptors that are activated by the binding of vitamin A derivatives and are involved in regulation of differentiation (see Chapter 30). Protein cofactors that interact with DNA-binding proteins but not the DNA itself further influence the rate of RNA synthesis by altering the three-dimensional arrangement of the general transcription apparatus at target gene promoters. These proteins are called transcriptional cofactors and serve as either coactivators or corepressors. The biological activity of some transcriptional cofactors is sensitive to nutritional status. For example, the peroxisome proliferator-activated receptor-gamma coactivator (PGC) 1 family of transcriptional cofactors are activated under conditions of nutrient deprivation. Expresson of PGC1 in liver increases with fasting and plays an important role in the regulation of gluconeogenesis by binding to and coactivating transcription factors such as hepatic nuclear factor (HNF) 4α, forkhead box class O (FoxO) 1, and the glucocorticoid receptor to coordinate the expression of rate-limiting gluconeogenic genes (Liang et al., 2009). Diverse protein–protein and protein–DNA combinations that result from transcriptional cofactors yield multiple layers of regulation and control, producing the phenotypic diversity seen among eukaryotic organisms and across tissues. Though this is important for development and coordination of complex biological systems, it also contributes to disease complexity and response variation to environmental stimuli. Newly transcribed pre-mRNA undergoes significant posttranscriptional processing as illustrated in Figure 13-6. First, the 5′ end of an eukaryotic mRNA is “capped” with a 7-methylguanosine residue (m7GTP). The covalently attached m7GTP molecule serves to protect the mRNA from exonucleases and more importantly is recognized by specific proteins of the translational machinery (Wilkie et al., 2003). Signals near the end of the template DNA strand denote the site for cleavage of the nascent pre-mRNA strand by an endonuclease and for polyadenylation at the 3′ end of the cut. All mature eukaryotic mRNAs, except histone mRNAs, have a 3′ poly(A) tail, which is a stretch of 20 to 250 adenosine residues added by polyadenylate polymerase. The poly(A) tail protects mRNA from degradation by exonucleases and serves as a binding region for poly(A)-binding protein, which functions in the circularization of mRNA during translation. The pre-mRNA also undergoes a process that excises the introns of the primary transcript and joins the exons to generate a mature mRNA product. This process of intron removal and exon ligation is called RNA splicing. It may begin before transcription of the gene is complete and must be completed before the mature mRNA is exported to the cytoplasm. Except for rare self-splicing introns, splicing requires a specialized RNA–protein complex called a spliceosome. Spliceosomes are multicomponent ribonucleoprotein complexes containing several small nuclear RNAs and more than 100 other proteins. This ribonucleoprotein complex assembles at the splice sites as the nascent pre-mRNA is transcribed. Mature mRNAs in association with ribonucleoproteins are exported through the nuclear pores into the cytoplasm. Differential processing of the pre-mRNA and variations in nucleocytoplasmic transport can affect the amount of mature mRNA in the cytoplasm. Incompletely processed mRNA is not exported from the nucleus. The level of the enzyme glucose-6-phosphate dehydrogenase (G6PD) is regulated at the level of pre-mRNA processing. G6PD is the first and rate-determining step of the pentose phosphate pathway, which serves an important role in carbohydrate metabolism and NADPH synthesis. Studies have shown that whereas the rate of G6PD transcription is constant irrespective of nutritional status (e.g., starvation versus refeeding), the amount of mature (spliced) G6PD mRNA is reduced during fasting and increased during refeeding. Splicing coactivator proteins modulate the efficiency of splicing of the nascent G6PD transcript, resulting in unspliced, partially spliced, and fully spliced forms. The balance of these forms influences the cytosolic G6PD mRNA level, and consequently G6PD protein level (Salati et al., 2004). A classic example of how mRNA stability is regulated through protein–mRNA interactions is found in iron metabolism. Transferrin receptor (TfR) mRNA expression is tightly linked to intracellular iron levels (Mullner and Kuhn, 1988). The 3′ UTR of TfR mRNA contains sequences called iron responsive elements (IrREs), which form stem–loop secondary structures that are susceptible to cleavage by RNA degrading enzymes (RNases). Under conditions of low iron, the stability of TfR mRNA is enhanced by the masking of these IrREs by iron regulatory proteins (IrRPs). Because the association of IrRPs with IrREs protects the mRNA from RNase cleavage, the transferrin receptor mRNA levels rise. Under conditions of high iron, the IrRE-binding activity of IrRPs is inactivated, allowing for increased TfR mRNA degradation. Many mRNAs contain AU-rich elements (AREs) that are targets of specialized RNA-binding proteins (ARE-RBPs). AREs are typically present in the 3′ UTR of mRNAs but are also found in 5′ UTR and in coding regions of some mRNAs. Some ARE-RBPs are stabilizing factors, whereas others are destabilizing factors. An example of this type of regulation is the regulation of renal glutaminase expression in response to acidosis (Ibrahim et al., 2008). The 3′ UTR of glutaminase mRNA contains AU sequences that function as pH-response elements to which several ARE-RBPs bind to variably stabilize or destabilize the glutaminase mRNA. When the pH of the cell drops, translocation of HuR (a stabilizing ARE-RBP) from the nucleus allows it to bind and stabilize glutaminase mRNA, resulting in increased glutaminase expression that enables ammonium ion production by the kidney for excretion of excess acid. Another common mechanism regulating mRNA stability is through the base pairing of miRNAs to the 3′ UTR of mRNA. Currently there are over 500 known mammalian genes that encode miRNAs, and each miRNA is capable of repressing hundreds of genes (Williams, 2008). Transcription of miRNA genes in the nucleus results in formation of primary miRNA, which are relatively short transcripts (~1 kb) that fold to form short hairpin structures. The primary miRNA is first processed in the nucleus by Drosha (a double-stranded RNA-specific ribonuclease) into a short hairpin structure called pre-miRNA. Following transport into the cytoplasm, pre-miRNA is further processed by a protein complex called Dicer, resulting in formation of short (20- to 30-nucleotide) RNA duplexes. After Dicer processing, the miRNA duplex is unwound and the mature miRNA strand binds to an argonaute protein to form the core component of the effector complex that mediates miRNA function. This complex is known as the RNA-induced silencing complex (RISC) (Pratt and MacRae, 2009). Usually only one strand of the mi-RNA duplex is loaded into the RISC complex; this tends to be the strand in which the 5′ end is less stably base-paired to its complement. The base pairing of miRNA within RISC to the 3′ UTR of a target mRNA promotes cleavage of the mRNA by ribonucleases, resulting in mRNA degradation. One of the four argonaute proteins in humans is an active endonuclease and can cleave mRNAs to which it binds with extensive complementarity. However, most miRNAs form base pairs with their mRNA targets with imperfect complementarity and repress the translation of their mRNA targets without endonucleolytic cleavage. Translational repression, however, commonly leads to mRNA destabilization and mRNA degradation by other machinery of the cell. The fact that miRNAs are small, can be rapidly transcribed, and do not need to be translated into protein to act gives miRNAs some advantages as regulators. Recent studies suggest that miRNAs may be important in the regulation of adaptive responses to nutritional stress or changes in environmental conditions (Strum et al., 2009). If the cellular abundance of all proteins was determined by the amount of mRNAs, the relationship between molar protein and mRNA levels would be linear. However, the correlation between mRNA levels and protein abundance in a single cell is poor, emphasizing the fact that changes in mRNA translation and protein turnover also play large roles in the regulation of cellular protein abundance. In the past decade, exploration regarding the control of gene expression at the level of mRNA translation has moved beyond uncovering basic details and toward development of novel means to treat and cure diseases such as cancer (Barnhart and Simon, 2007). Ribosome biogenesis describes the making and assembling of ribosomal proteins and rRNAs into the 40S and 60S ribosomal subunits. Expression of the genes encoding the numerous constituents of ribosomes requires transcription by all three classes of RNAPs (Mayer and Grummt, 2006). A signaling network in yeast named target of rapamycin (TOR) is identified as critical in controlling ribosomal protein gene expression and coordinating the relative activity of all three RNAPs to achieve the proper stoichiometry of ribosomal components. Both nutrients and stress can influence mammalian TOR (mTOR) signaling, linking ribosomal capacity to nutrient availability and other environmental cues. Conditions of rapid growth require enhanced ribosome production, and increased levels of ribosomes have been observed in growing tumors (Belin et al., 2009). Ribosomal protein mRNAs all contain a cis-regulatory element consisting of several pyrimidines at the 5′ end. This terminal oligopyrimidine (TOP) tract is also present in mRNA translation elongation factors and poly(A)-binding protein. Feeding a protein-containing meal maximally enhances TOP mRNA translation, whereas amino acid deficiency completely abrogates translation of TOP mRNAs (Anthony et al., 2001). This exaggerated “all-or-none” binary control mechanism suggests that in the repressed state, translation is blocked. A number of studies have implicated the phosphatidylinositol 3-kinase (PtdIns3K) and mTOR signaling pathways in the activation of TOP mRNAs during high growth conditions, but consensus on the mechanism underlying this regulatory process has not been reached. The regulation of translational capacity provides the organism with an ability to adapt to chronic or sustained conditions of change. The majority of translational control lies at the initiation step, which is summarized in Figure 13-7. This step can be further subdivided into three events that determine overall initiation activity. The first event involves assembly of a ternary complex (TC) consisting of the initiating tRNA (specifically, a particular initiator methionyl-tRNA, or Met-tRNAi) bound to the protein factor eIF2 in association with GTP (guanosine 5′-triphosphate). eIF2 is a guanine nucleotide-binding protein (i.e., G-protein) made of α, β, and γ subunits. eIF2 exists either in an active GTP-bound state or in an inactive GDP (guanosine 5′-diphosphate)-bound form. Only when eIF2 is in the GTP-bound form can the TC bind the small ribosomal subunit. Following TC formation, the TC and other protein factors (e.g., eIF1, eIF3, eIF5) bind to the 40S ribosomal subunit to form the 43S preinitiation complex and eIF2–GDP is released (Lorsch and Dever, 2010). Regeneration of the eIF2–GTP from eIF2–GDP is catalyzed by eIF2B, a guanine nucleotide exchange factor (GEF). The GEF activity of eIF2B is regulated primarily by phosphorylation of eIF2 on its α subunit, which increases its binding affinity to several eIF2B subunits, stalling eIF2B GEF activity. Phosphorylation of eIF2 is catalyzed by a family of four kinases, each responsive to a distinct set of environmental stressors. The mammalian eIF2α kinases are heme-regulated inhibitor (HRI), which is sensitive to heme deprivation; double-stranded RNA-dependent protein kinase (PKR), which is activated by a viral infection; PKR-like endoplasmic reticulum resident kinase (PERK), which is activated by misfolded proteins or other stress in the ER; and general control nonderepressible kinase 2 (GCN2), which is activated by conditions of amino acid deprivation (Wek et al., 2006). Another way in which the GEF activity of eIF2B is modulated is by the binding of the protein factor eIF5 to eIF2, sequestering eIF2 away from eIF2B, thus preventing guanine nucleotide exchange (Singh et al., 2006). The second event in translation initiation subject to major regulation involves the binding of the 43S preinitiation complex to the selected mRNA. This event requires eIF3 and several other initiation factors collectively called eIF4 (or eIF4F). One of the proteins in this group, named eIF4E, selects the mRNA to be translated by binding its 5′-m7GTP cap structure. A second member of the eIF4 group, called eIF4G, functions as a scaffold to bring the small ribosomal subunit and the mRNA close to each other. eIF4G accomplishes this task by binding both eIF4E, which is bound to the mRNA cap, and eIF3, which is associated with the 40S ribosomal subunit in the preinitiation complex. A family of repressor proteins known as the eIF4E-binding proteins (4E-BPs) can prevent the interaction of eIF4G and eIF4E and thereby inhibit the 40S ribosome from binding mRNA. The repressor activity of the 4E-BP is regulated by phosphorylation, with increased phosphorylation reducing its affinity to associate with eIF4E. A second function of eIF4G is to associate with poly(A)-binding protein, which results in 5′,3′-circularization of mRNA, as shown in the lower part of Figure 13-7. Circularization of mRNA is believed to be important for stabilizing recruited 40S ribosomal subunits and for efficient recycling of terminating ribosomes for another round of translation of the same mRNA (Wilkie et al., 2003). After the eIF4 complex has brought the 43S preinitiation complex and mRNA together, the small ribosomal subunit moves along the mRNA toward the 3′ end scanning for the start codon. Scanning is facilitated by eukaryotic initiation factor eIF4A, which functions as an ATP-dependent helicase to unwind mRNA secondary structure in the 5′ UTR. Start codon recognition halts scanning and triggers a number of events that commit the ribosome to begin translation at that point on the mRNA. eIF1, eIF2, eIF5, and eIF5B play key roles in these events. First, eIF1 is released from the small ribosomal subunit, denoting recognition of the correct AUG start codon (Cheung et al., 2007). In addition, upon AUG recognition, GTP hydrolysis on eIF2 is triggered, an event aided by eIF5. The final event involves eIF5B-mediated joining of the 60S large ribosomal subunit with the 40S small ribosomal subunit on the mRNA to form an 80S ribosome along with release of eIF2–GDP and other initiation factors. eIF2–GDP must be converted back to eIF2–GTP before it can participate in another round of initiation. After initiation, the polypeptide is assembled with the amino acid sequence being specified by the mRNA sequence, as illustrated in Figure 13-8. This process requires substantial metabolic energy, with two molecules of GTP cleaved for every added amino acid. The elongation step of mRNA translation involves fewer protein factors than the initiation step, but the eEFs required are considered the workhorses of protein synthesis on the ribosome. During initiation, the Met-tRNAi is base-paired with the mRNA start codon in a location named the peptidyl or “P” site within the ribosome. The protein factor eEF1A with bound GTP then delivers the next correct aminoacyl–tRNA to the ribosome at the aminoacyl or “A” site, beginning the process of elongation. Upon correct codon–anticodon interaction, GTP is hydrolyzed and eEF1A with bound GDP is released from the ribosome. A second factor, a GEF named eEF1B, assists in regenerating active eEF1A–GTP, ensuring continued deliverance of aminoacyl–tRNAs to the ribosome. With delivery of an aminoacyl–tRNA into the A site, peptide bond formation proceeds, joining the amino group of the aminoacyl–tRNA in the A site to the carboxyl group of the aminoacyl–tRNA in the P site with release of water. Peptide bond synthesis is catalyzed by a peptidyl transferase that is part of the 60S ribosomal subunit. A third and final factor, named eEF2, facilitates movement of the ribosome along the mRNA, resulting in translocation of the peptidyl–tRNA from the A site to the P site and movement of the unloaded tRNA in the P site to the “E site,” where it can exit the ribosome. The energy required for the ribosome to rachet forward on the mRNA is provided by GTP hydrolysis, catalyzed by eEF2. A GEF is not needed to regenerate eEF2–GTP because eEF2 has low affinity for GDP which spontaneously dissociates. All three eEFs are subject to phosphorylation in mammalian cells. Phosphorylation of eEF1A stimulates elongation activity, whereas phosphorylation of its GEF, eEF1B, on several subunits has no reported influence on elongation rates. On the other hand, eEF2 is inactivated by phosphorylation. Phosphorylation of eEF2 does not impact the ability of the ribosome to hydrolyze GTP but instead reduces the affinity of eEF2 for the ribosome (Carlberg et al., 1990). Phosphorylation of eEF2 occurs in response to stimuli that either increase energy demand or reduce its supply. This likely serves to slow down protein synthesis and thus conserve energy under such circumstances (Andersen et al., 2003). Examples of conditions that increase eEF2 phosphorylation include intense exercise, alcohol intake, ischemia, and denervation. The N-terminal methionine residue is removed from many proteins, and other proteins are cleaved to produce active peptides, remove targeting sequences that are no longer needed, or remove inhibitory prosequences for activation of the protein. Other common modifications include, but are not limited to, disulfide bond formation, glycosylation, acetylation, and fatty acylation. Some proteins require the addition of a cofactor such as heme or biotin. Despite the widespread presence of protein modifications in nature, much remains unknown about how these alterations affect the activity of each individual protein. This is mostly because a singular type of modification imparts different functions, depending on the target protein. For example, N-linked glycosylation may stabilize the folding process of one polypeptide, allow another protein to play an important role in cell–cell recognition, or protect a lysosomal membrane protein from degradation by lysosomal proteases. The mechanisms for these and other protein modifications during and following translation is varied and complex. Mutations in genes encoding proteins involved in protein modifications are among the causes of inherited disease. For example, mutations in several genes encoding proteins involved in the glycosylation of alpha-dystroglycan have been identified as a cause of one group of muscular dystrophies (Muntoni et al., 2008).
Protein Synthesis and Degradation
Protein Turnover
Overview of Protein Turnover
Synthesis and Degradation of Protein in Relation to Protein Balance
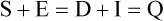
Protein Turnover and Adaptation
ENZYME
CELLULAR COMPARTMENT
t½
kd (% per day)
Ornithine decarboxylase
Cytosol
11 minutes
91
5-Aminolevulinate synthetase
Cytosol
20 minutes
50
5-Aminolevulinate synthetase
Mitochondria
72 minutes
14
Hydroxymethylglutaryl CoA reductase
Endoplasmic reticulum
4.0 hours
4.2
Phosphoenolpyruvate carboxykinase
Cytosol
5.0 hours
3.3
Alanine-glyoxylate aminotransferase
Cytosol
3.5 days
0.20
Arginase
Cytosol
4.0 days
0.17
NAD+ nucleosidase
Endoplasmic reticulum
16 days
0.04
Protein Synthesis
DNA Transcription
RNA Polymerases
Regulation of Transcription by Transcription Factors
Regulation of Transcription by Coactivators and Corepressors
Pre-mRNA Processing and Export
Stability of mRNA
Regulation of mRNA Stability by RNA-Binding Proteins
Regulation of mRNA Stability by miRNA
Translation of mRNA
Regulation of Translation by Regulation of Ribosome Biogenesis
Translational Efficiency
Control of Translation Initiation: Formation of Preinitiation Complex and Its Regulation by eIF2α Phosphorylation
Control of Translation Initiation: Association of the 40S Ribosomal Subunit with mRNA by Cap-Dependent Recognition and Its Regulation by eIF4E Binding Protein Phosphorylation
Start Codon Recognition, Large Subunit Engagement, and eIF2 Recycling
Polypeptide Synthesis: Elongation and Its Regulation
Posttranslational Modifications and Folding of Proteins
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine