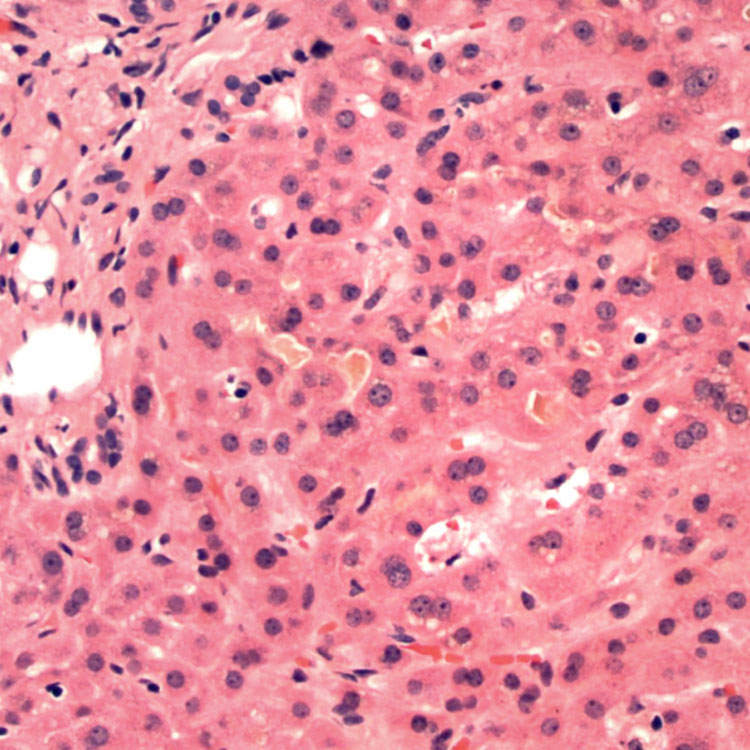
Giant Cell Transformation H&E-stained section of a liver biopsy in a child with progressive familial intrahepatic cholestasis (PFIC) shows giant cell transformation of perivenular hepatocytes, typical of childhood cholestasis syndromes.
Bland Canalicular Cholestasis H&E-stained section of a liver biopsy in an adult patient with benign recurrent intrahepatic cholestasis (BRIC) shows bland canalicular cholestasis with mild lobular architectural disarray but minimal inflammation.
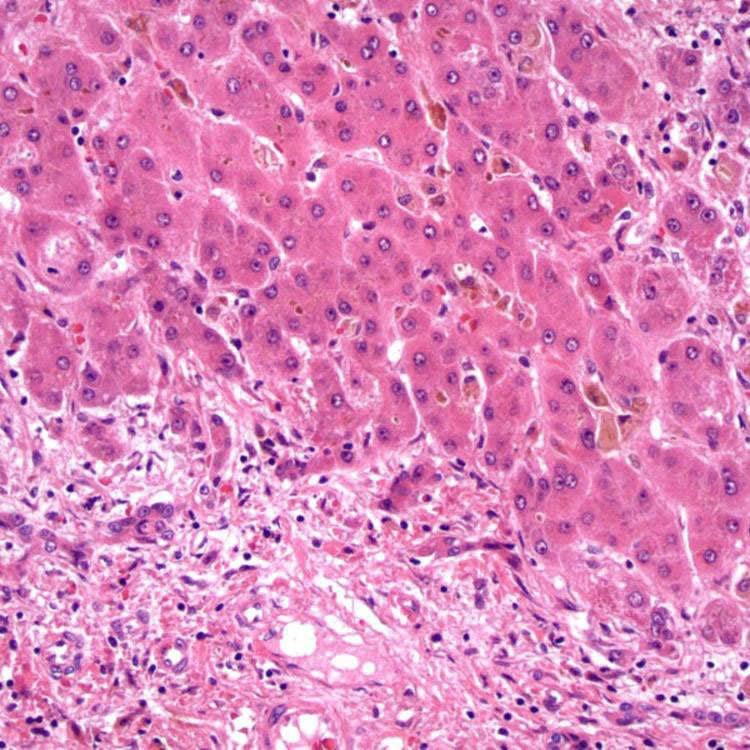
Periportal Cholestasis H&E-stained section of a liver biopsy in a patient with PFIC1 shows periportal cholestasis and bile ductular reaction in the portal tract.
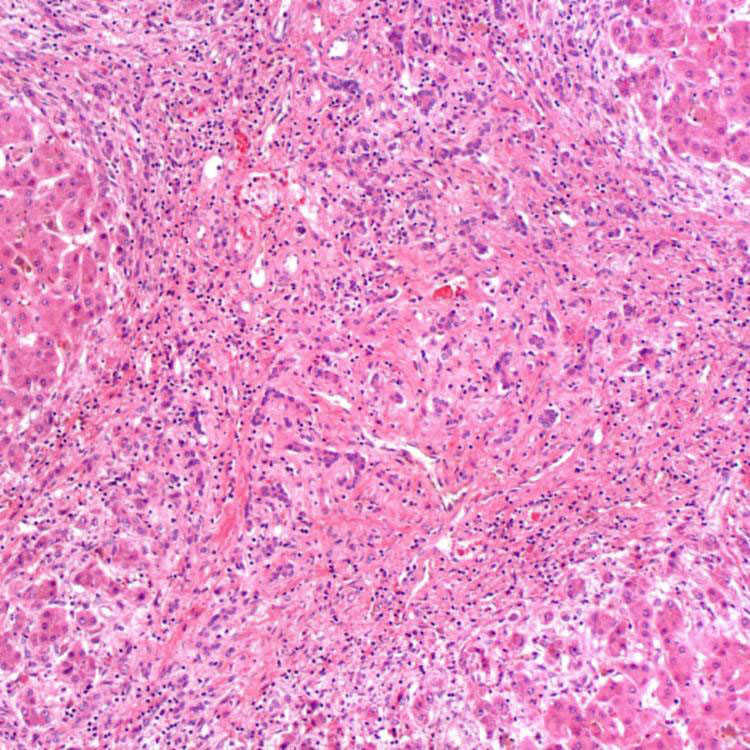
Fibrous Septa H&E-stained section of a liver explant in a patient with PFIC1 shows periseptal cholestasis, intrahepatocytic clearing, and bile ductular reaction in the fibrous septa.
• Heterogeneous group of autosomal recessive disorders characterized by chronic cholestasis and progression to cirrhosis and liver failure
ETIOLOGY/PATHOGENESIS
Autosomal Recessive Genetic Disorder
• PFIC1
Mutation of ATP8B1 (FIC1) gene, located on chromosome 18q21-q22
– FIC1 is expressed on variety of tissues including liver, intestine, pancreas
– Functions as aminophospholipid flippase, flipping phosphatidylserine from outer to inner lipid layer of cell membrane
– Mechanism of cholestasis unclear
• PFIC2
Mutations of ABCB11 gene on chromosome 2q24 that encodes BSEP, an ATP-dependent bile acid transporter on canalicular membrane
• PFIC3
Mutation of ABCB4 gene that encodes MDR3 glycoprotein
– MDR3 is flippase that flips phosphatidylcholine from inner to outer lipid leaflet of canalicular membrane
– Phosphatidylcholine in bile reduces its detergent action, and MDR3 deficiency results in bile with more detergent properties
– Absence of phospholipids destabilizes micelles, promoting lithogenicity of bile with crystallization of cholesterol and leads to small bile duct obstruction
CLINICAL ISSUES
Presentation
• FIC1 deficiency disease
Depending on nature of mutation, may present as benign recurrent intrahepatic cholestasis (BRIC1) or progressive and severe form (PFIC1)
PFIC1
– Presents in 1st year of life with intense pruritus and jaundice
– Systemic disorder with extrahepatic manifestations including pancreatitis, diarrhea, respiratory symptoms, failure to thrive, delayed sexual development, hearing loss
BRIC1
– Recurrent episodes of cholestasis with intense pruritus
– Episodes resolve spontaneously without histologic progression
• BSEP disease
Depending on nature of mutation, may present as BRIC2 or PFIC2
PFIC2
– Presents as severe intrahepatic cholestasis in infancy
BRIC2
– Presents as recurrent episodes of pruritus, steatorrhea, nausea, vomiting, anorexia, right upper quadrant abdominal pain, and weight loss
– Frequently complicated by cholesterol cholelithiasis
• MDR3 disease
PFIC3 presents during infancy with pruritus, jaundice, pale stools, hepatomegaly, or complications of portal hypertension, such as splenomegaly or gastrointestinal bleeding
MDR3 mutations also seen in patients with intrahepatic lithiasis, cholesterol gallstone disease, intrahepatic cholestasis of pregnancy, transient neonatal cholestasis, cholestatic drug reactions
Laboratory Tests
• GGT
Normal in PFIC1 and PFIC2
Elevated in PFIC3
• Elevated serum bile acids in all 3 types
• PFIC3 is characterized by low concentrations of phospholipids in bile analysis
Natural History
• Progressive forms can result in worsening hepatic function, liver failure, cirrhosis, and death before adulthood
Chronic cholestasis leads to complications of fat malabsorption such as deficiencies of fat-soluble vitamins and weight loss
Only gold members can continue reading. Log In or Register to continue














 PFIC3 presents during infancy with pruritus, jaundice, pale stools, hepatomegaly, or complications of portal hypertension, such as splenomegaly or gastrointestinal bleeding
PFIC3 presents during infancy with pruritus, jaundice, pale stools, hepatomegaly, or complications of portal hypertension, such as splenomegaly or gastrointestinal bleeding


 Chronic cholestasis leads to complications of fat malabsorption such as deficiencies of fat-soluble vitamins and weight loss
Chronic cholestasis leads to complications of fat malabsorption such as deficiencies of fat-soluble vitamins and weight loss




