27.1 INTRODUCTION TO PRIONS
This chapter is not about viruses, but about some infectious diseases of man and animals that are known as prion diseases. Much of the research on prions is published in virology journals; hence their appearance in this book. The evidence suggests that the causative agents are protein molecules from within the cells of the host; no nucleic acid has been found associated with them.
The prion diseases are characterized by very long incubation periods, measured in years. (The incubation period of an infectious disease is the time that elapses between the infectious agent entering the host and the first appearance of signs and symptoms of the disease.) Signs of prion disease include dementia and loss of coordination; the patient gradually deteriorates, and death is inevitable.
This subject assumed particular importance toward the end of the twentieth century with the onset in the UK of bovine spongiform encephalopathy (BSE), or mad cow disease as it is popularly known, and its apparent transmission to humans as variant Creutzfeldt-Jakob disease (vCJD).
27.2 TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES
The prion diseases are known as transmissible spongiform encephalopathies (TSEs). Taking these words in reverse order:
- encephalopathy means disease of the brain;
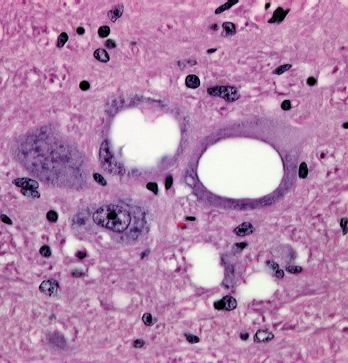
- spongiform refers to the development of holes in the brain, making it appear like a sponge (Figure 27.1);
Figure 27.1 Brain section from a sheep with scrapie. The spongiform appearance (holes in the tissue) is evident. Magnification × 500.
Source: Courtesy of Dr R. Higgins, University of California, Davis.
- transmissible refers to the fact that the causative agent is infectious. It can be transmitted to members of the same species, and sometimes to other species.
27.3 THE NATURE OF PRIONS
The agents that cause TSEs appear to be misfolded forms of normal cell proteins; there is no evidence that they contain nucleic acid. This “protein-only” hypothesis was proposed by Stanley Prusiner, who also suggested the term prion (pronounced pree-on), derived from infectious protein. Versions of the normal protein have been found in mammals, birds, reptiles, and fish; in humans it is encoded by the PRNP gene. The role of the protein is not yet clear. It cycles between endosomes and the cell surface, where it is held in the plasma membrane by a glycosyl-phosphatidyl-inositol anchor at its C terminus. It is found on many cell types, but especially on cells of the central nervous system.
The prion protein molecule has a loop as a result of a disulfide bond (Figure 27.2). The loop contains two asparagine residues, either or both of which can be N-glycosylated, so the protein exists as three glycoforms (unglycosylated, monoglycosylated, and diglycoslyated); the sugars attached to the protein may be of various types.
Figure 27.2 Normal and misfolded forms of prion. (a) and (b) representations of the normal form; (c) the misfolded form has more β sheet (shown in green) than the normal form.
Source: (b) and (c) from Cervia et al. (2006) Transfusion Medicine Reviews, 20, 190. Reproduced by permission of Elsevier.
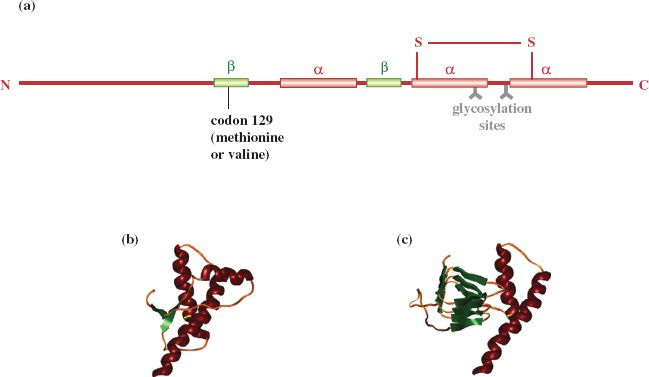
The conformation of much of the normal protein is α helix (Figure 27.2). When the protein misfolds there is a decrease in α helix structure and an increase in β sheet. This change in conformation is accompanied by changes in properties of the protein, including susceptibility to digestion by proteinase K. The normal protein is completely digested, but the misfolded protein is largely resistant to the enzyme, which is able to digest only 90–100 amino acid residues from the N terminus, the remaining 27–30 kD portion of the protein remaining intact. Misfolding also renders the protein insoluble in nonionic detergents. In prion-diseased tissues molecules of the misfolded protein aggregate as fibrils, rods, or other forms, depending on the host and the prion strain.
Various terminologies are used for the normal and the misfolded forms of the protein. The normal protein is commonly designated as PrPc (PrP=prion protein; c=cell), while the misfolded form is designated as PrPSc (Sc=scrapie) or PrPres (res=resistant to proteinase).
The presence of certain amino acids at particular sites in the prion protein can affect a number of aspects of prion disease, including susceptibility to prion diseases and incubation period. For example, the PRNP gene encodes either methionine or valine at codon 129 (Figure 27.2). There can be very different outcomes depending whether the individual is homozygous (methionine/methionine or valine/valine) or heterozygous (methionine/valine). So far, all cases of vCJD have been homozygous for methionine at codon 129. Small variations in the prion gene sequence can also strongly influence the clinical course of a prion disease.
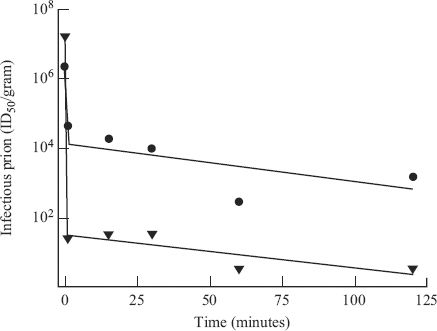
Prion infectivity is remarkably heat resistant and some infectivity can survive autoclaving for prolonged periods (Figure 27.3). It is interesting to note that a small subpopulation of the infective material has a higher level of resistance to inactivation. This subpopulation is inactivated at a slower rate, mirroring the situation with many viruses (Section 24.4). Prion infectivity is also very resistant to inactivation by irradiation and by some chemicals that inactivate virus infectivity. Treatments that are used to inactivate prion infectivity include exposure to 5% sodium hypochlorite solution or 1 M NaOH for 1–2 hours.
Figure 27.3 Inactivation of two strains of scrapie prion in an autoclave at 126 °C.
Source: Data from Somerville (2002) Trends In Biochemical Sciences, 27, 606. Reprinted by permission of Elsevier and the author.
27.3.1 Prion replication
The mechanism by which prion “replication” takes place is not fully understood. It has been suggested that a form of “evangelism” is involved, whereby molecules of the misfolded protein “convert” normal protein molecules, causing them to misfold. In other words, the misfolded protein causes a change in the conformation of the normal protein.
A further suggestion is that initiation of the replication process requires a “seed” consisting of an aggregate of misfolded protein molecules. There are three ways in which this “seed” could arise: it could be introduced into the body, it could be formed after a rare conformational change in a normal protein molecule, or it could be the inevitable outcome of the expression of a mutant prion gene.
The formation of an infectious pathogenic agent as a result of host protein molecules undergoing a conformational change is a very different process to virus replication, where new virions are constructed from amino acids and nucleotides. It has been difficult for many to accept the idea that an infectious agent that is able to replicate is devoid of any nucleic acid, but all attempts to demonstrate the presence of a nucleic acid have failed.
The following work, published in 2004, provides evidence that prions are infectious proteins. Mouse prion protein was produced in recombinant bacterial cells and polymerized into fibrils, which were injected into the brains of mice. The mice subsequently developed signs of a TSE and extracts of their brains transmitted the disease to other mice.
The misfolded protein accumulates in endosomes and lysosomes, and quantities build up especially in neurons. In some prion diseases the misfolded protein can also be found in a number of other organs and tissues, including the spleen and lymph nodes.
27.4 PRION DISEASES
Some cases of prion disease arise spontaneously, some are inherited and some are acquired as a result of a TSE agent entering the body. In the latter case entry may be through the digestive tract, followed by transport to the lymphoreticular system (e.g. spleen, lymph nodes, Peyer’s patches), where the molecules are amplified. From these sites they can be transferred into the central nervous system.
Replication takes place slowly but, because the misfolded protein is not degraded, concentrations gradually build up. The molecules form insoluble aggregates, often visible as “plaques” in sections of central nervous tissue. The accumulation of these aggregates is thought to lead to dysfunction and death of neurons, leading to the development of the holes in the brain (Figure 27.1). The inevitable outcome is the death of the host.
27.4.1 Prion diseases in animals
Scrapie is a disease of sheep and goats that has been known in Britain and other parts of Europe for hundreds of years. Many affected animals scrape against hard objects such as fence posts, hence the name of the disease, and many grind their teeth, stumble, and fall; all eventually die. In the 1930s it was demonstrated that scrapie can be transmitted from sheep to sheep by injection of brain tissue. It was mentioned earlier that small variations in the prion gene sequence can affect susceptiblity to infection. It has been found that sheep are more susceptible to scrapie if the prion gene has valine rather than alanine at codon 136.
More recently a distinct TSE has been reported in sheep and is referred to as atypical scrapie.
In the US a TSE in farmed mink was first recognized in 1947, then in the early 1980s chronic wasting disease was described in mule deer and elk in captivity. The latter disease has since been found in wild mule deer, white-tailed deer, elk, and moose; it is the only TSE known to occur in free-ranging animals.
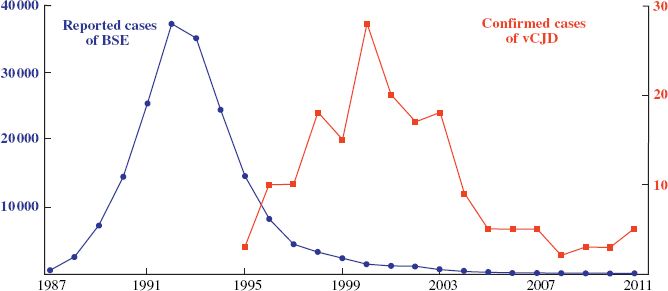
BSE was first reported in the UK in 1986 and a massive outbreak rapidly developed (Figure 27.4). The disease was spread by feeding meat and bone meal to cattle as a protein supplement. Body parts from animals incubating the disease were incorporated into meat and bone meal and this served to spread the disease on a large scale. Cattle that appeared to be healthy could in fact be incubating BSE and there could be large quantities of misfolded protein in their brains and spinal cords. It is uncertain whether the original infectious material was from a sheep with scrapie or from a cow in which BSE had arisen spontaneously.
Figure 27.4 Numbers of cases of BSE and vCJD in the UK.
Source: BSE data from World Organization for Animal Health. vCJD data from UK National CJD Surveillance Unit.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


