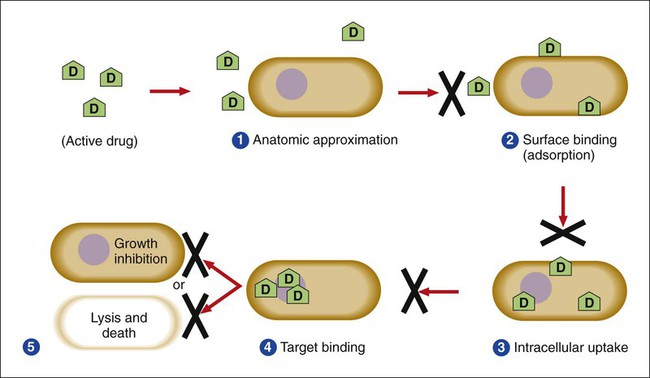
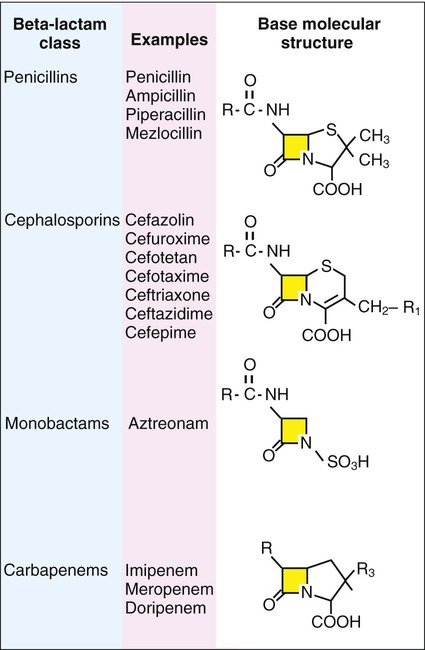
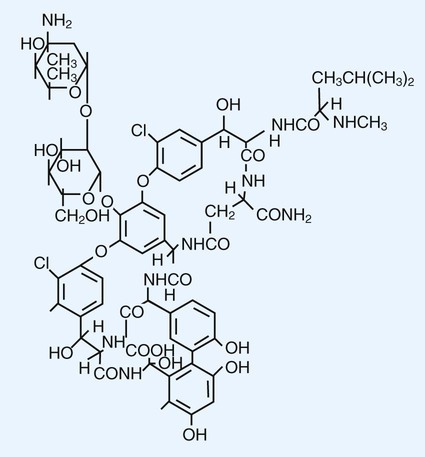
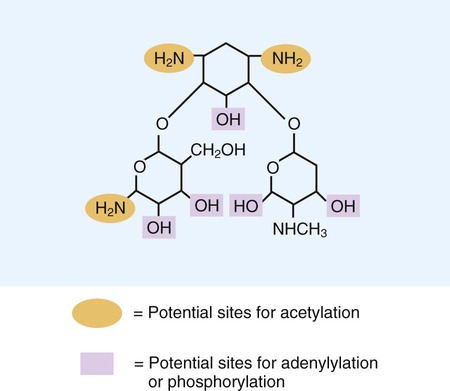
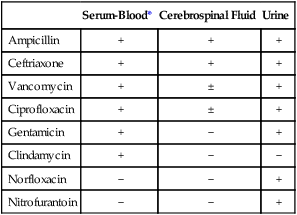
1. List the five general categories of antimicrobial actions. 2. Define antibiotic and antimicrobial. 3. Define and differentiate bactericidal and bacteriostatic agents. 4. Compare and contrast the following terms: biologic versus clinical resistance, environmentally mediated versus microorganism-mediated resistance and intrinsic versus acquired resistance. 5. Describe the basic structure and chemical principle for the mechanism of beta-lactam antimicrobials. 6. List common β-lactam antibiotics and provide an example of a common pathogen susceptible to these agents 7. Describe the chemical principle for the mechanism of resistance to β-lactam antibiotics. 8. Describe the chemical principle for the mechanisms of glycopeptide agents. 9. List common glycopeptides and provide an example of a common pathogen susceptible to these agents. 10. List examples of cell membrane inhibitors, inhibitors of protein synthesis, inhibitors of deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) synthesis, and metabolic inhibitors. Provide an example of a common pathogen susceptible to each of the agents listed. 11. List five general mechanisms for antimicrobial resistance and provide an example for each. 12. Describe how the dissemination of antimicrobial resistance affects diagnostic microbiology, including effects on sensitivity testing, therapeutics, and organism identification. Because antimicrobial agents play a central role in the control and management of infectious diseases, understanding their mode of action and the mechanisms of microorganisms to circumvent antimicrobial activity is important, especially because diagnostic laboratories are expected to design and implement tests that measure a pathogen’s response to antimicrobial activity (see Chapter 12). Much of what is discussed here regarding antimicrobial action and resistance is based on antibacterial agents, but the principles generally apply to almost all antiinfective agents. More information about antiparasitic, antifungal, and antiviral agents can be found in Parts IV, V, and VI, respectively. Several key steps must be completed for an antimicrobial agent to successfully inhibit or kill an infecting microorganism (Figure 11-1). First, the agent must be in an active form. This is ensured through the pharmacodynamic design of the drug, which takes into account the route by which the patient receives the agent (e.g., orally, intramuscularly, intravenously). Second, the antibiotic must also be able to achieve sufficient levels or concentrations at the site of infection so that it has a chance to exert an antibacterial effect (i.e., it must be in anatomic approximation with the infecting bacteria). The ability to achieve adequate levels depends on the pharmacokinetic properties of the agent, such as rate of absorption, distribution, metabolism, and excretion of the agent’s metabolites. Table 11-1 provides examples of various anatomic limitations characteristic of a few commonly used antibacterial agents. Some agents, such as ampicillin and ceftriaxone, achieve therapeutically effective levels in several body sites, whereas others, such as nitrofurantoin and norfloxacin, are limited to the urinary tract. Therefore, a knowledge of the site of infection can substantially affect the selection of the antimicrobial agent for therapeutic use. TABLE 11-1 Anatomic Distribution of Some Common Antibacterial Agents +, Therapeutic levels generally achievable at that site; ±, therapeutic achievable levels moderate to poor; −, therapeutic levels generally not achievable at that site. The remaining steps in antimicrobial action relate to direct interactions between the antibacterial agent and the bacterial cell. The antibiotic is attracted to and maintains contact with the cell surface. Because most targets of antibacterial agents are intracellular, uptake of the antibiotic to some location inside the bacterial cell is required. Once the antibiotic has achieved sufficient intracellular concentration, binding to a specific target occurs. This binding involves molecular interactions between the antimicrobial agent and one or more biochemical components that play an important role in the microorganism’s cellular metabolism. Adequate binding of the target results in disruption of cellular processes, leading to cessation of bacterial cell growth and, depending on the antimicrobial agent’s mode of action, cell death. Antimicrobial agents that inhibit bacterial growth but generally do not kill the organism are known as bacteriostatic agents. Effectively reducing the growth rate of an organism provides adequate protection in individuals whose immune system is capable of removing the agent of infection. Agents that usually kill target organisms are said to be bactericidal (Box 11-1). Bacteriocidal agents are more effective against organisms that are more difficult to control in combination with the host’s immune system. The primary goal in the development and design of antimicrobial agents is to optimize a drug’s ability to efficiently achieve all steps outlined in Figure 11-1 while minimizing toxic effects on human cells and physiology. Different antibacterial agents exhibit substantial specificity in terms of their bacterial cell targets, that is, their mode of action. For this reason, antimicrobial agents are frequently categorized according to their mode of action. The interior of the bacterial cell has several potential antimicrobial targets. However, the processes or structures most frequently targeted are cell wall (peptidoglycan) synthesis, the cell membrane, protein synthesis, metabolic pathways, and DNA and RNA synthesis (Table 11-2). TABLE 11-2 Summary of Mechanisms of Action for Commonly Used Antibacterial Agents β-lactam antibiotics have a four-member, nitrogen-containing, β-lactam ring at the core of their structure (Figure 11-2). The antibiotics differ in ring structure and attached chemical groups. This drug class comprises the largest group of antibacterial agents, and dozens of derivatives are available for clinical use. Types of β-lactam agents include penicillins, cephalosporins, carbapenems, and monobactams. The popularity of these agents results from their bactericidal action and lack of toxicity to humans; also, their molecular structures can be manipulated to achieve greater activity for wider therapeutic applications. To circumvent the development of antimicrobial resistance, β-lactam combinations comprised of a β-lactam with antimicrobial activity (e.g., ampicillin, amoxicillin, piperacillin) and a beta-lactam without activity capable of binding and inhibiting β-lactamases (e.g., sulbactam, clavulanate, tazobactam) have been developed. The binding β-lactam “ties up” the β-lactamases produced by the bacteria and allows the other β-lactam in the combination to exert its antimicrobial effect. Examples of these β-lactam/β-lactamase inhibitor combinations include ampicillin/sulbactam, amoxicillin/clavulanate, and piperacillin/tazobactam. Such combinations are effective only against organisms that produce β-lactamases that are bound by the inhibitor; they have little effect on resistance that is mediated by altered PBPs (see Mechanisms of Antibiotic Resistance later in this chapter). Glycopeptides are the other major class of antibiotics that inhibit bacterial cell wall synthesis by binding to the end of the peptidoglycan, interfering with transpeptidation. This is a different mechanism from that of the β-lactams, which bind directly to the enzyme. Two such antibiotics, vancomycin and teicoplanin, are large molecules and function differently from β-lactam antibiotics (Figure 11-3). With glycopeptides, the binding interferes with the ability of the PBP enzymes, such as transpeptidases and transglycosylases, to incorporate the precursors into the growing cell wall. With the cessation of cell wall synthesis, cell growth stops and death often follows. Because glycopeptides have a different mode of action, the resistance to β-lactam agents by gram-positive bacteria does not generally hinder their activity. However, because of their relatively large size, they cannot penetrate the outer membrane of most gram-negative bacteria to reach their cell wall precursor targets. Therefore, this agent is usually ineffective against gram-negative bacteria. Teicoplanin is approved for use throughout the world but is not currently available in the United States. When vancomycin is used, its levels should be monitored because the potential for toxicity. Aminoglycosides (aminoglycosidic aminocyclitol) inhibit bacterial protein synthesis by irreversibly binding to protein receptors on the organism’s 30S ribosomal subunit. This process interrupts several steps, including initial formation of the protein synthesis complex, accurate reading of the messenger RNA (mRNA) code, and formation of the ribosomal-mRNA complex. The structure of a commonly used aminoglycoside, gentamicin, is shown in Figure 11-4. Other aminoglycosides include tobramycin, amikacin, streptomycin, and kanamycin. The spectrum of activity of aminoglycosides includes a wide variety of aerobic gram-negative and certain gram-positive bacteria, such as S. aureus. Bacterial uptake of the aminoglycosides is accomplished by using them in combination with cell wall–active antibiotics, such as β-lactams or vancomycin. Anaerobic bacteria are unable to uptake these agents intracellularly and therefore are typically not inhibited by aminoglycosides. Aminoglycosides are associated with toxicity, and blood levels should be monitored during therapy. The major toxicities are nephrotoxicity and auditory or vestibular toxicity.
Principles of Antimicrobial Action and Resistance
Antimicrobial Action
Principles
Serum-Blood*
Cerebrospinal Fluid
Urine
Ampicillin
+
+
+
Ceftriaxone
+
+
+
Vancomycin
+
±
+
Ciprofloxacin
+
±
+
Gentamicin
+
−
+
Clindamycin
+
−
−
Norfloxacin
−
−
+
Nitrofurantoin
−
−
+
Mode of Action of Antibacterial Agents
Antimicrobial Class
Mechanism of Action
Spectrum of Activity
Aminoglycosides (e.g., gentamicin, tobramycin, amikacin, streptomycin, kanamycin)
Inhibit protein synthesis by binding to 30S ribosomal subunit
Gram-positive and gram-negative bacteria; not anaerobic bacteria
β-lactams (e.g., penicillin, ampicillin, mezlocillin, piperacillin, cefazolin, cefotetan, ceftriaxone, cefotaxime, ceftazidime, aztreonam, imipenem)
Inhibit cell wall synthesis by binding enzymes involved in peptidoglycan production (i.e., penicillin-binding proteins [PBPs])
Both gram-positive and gram-negative bacteria, but spectrum may vary with the individual antibiotic.
Chloramphenicol
Inhibits protein synthesis by binding 50S ribosomal subunit
Gram-positive and gram-negative bacteria
Fluoroquinolones (e.g., ciprofloxacin, ofloxacin, norfloxacin)
Inhibit DNA synthesis by binding DNA gyrase and topoisomerase IV
Gram-positive and gram-negative bacteria, but spectrum may vary with individual antibiotic
Glycylglycines (e.g., tigecycline)
Inhibition of protein synthesis by binding to 30S ribosomal subunit
Wide spectrum of gram-positive and gram-negative species including those resistant to tetracycline
Ketolides (e.g., telithromycin)
Inhibition of protein synthesis by binding to 50S ribosomal subunit
Gram-positive cocci including certain macrolide-resistant strains and some fastidious gram-negatives (e.g., H. influenzae and M. catarrhalis)
Lipopeptides (e.g., daptomycin)
Binding and disruption of cell membrane
Gram-positive bacteria including those resistant to beta-lactams and glycopeptides
Nitrofurantoin
Exact mechanism uncertain; may have several bacterial enzyme targets and directly damage DNA
Gram-positive and gram-negative bacteria
Oxazolidinones (e.g., linezolid)
Bind to 50S ribosomal subunit to interfere with initiation of protein synthesis
Wide variety of gram-positive bacteria, including those resistant to other antimicrobial classes
Polymyxins (e.g., polymyxin B and colistin)
Disruption of cell membrane
Gram-negative bacteria, poor activity against most gram-positive bacteria
Rifampin
Inhibits RNA synthesis by binding DNA-dependent, RNA polymerase
Gram-positive and certain gram-negative (e.g., N. meningitidis) bacteria
Streptogramins (e.g., quinupristin/dalfopristin)
Inhibit protein synthesis by binding to two separate sites on the 50S ribosomal subunit
Primarily gram-positive bacteria
Sulfonamides
Interfere with folic acid pathway by binding the enzyme dihydropteroate synthase
Gram-positive and many gram-negative bacteria
Tetracycline
Inhibits protein synthesis by binding 30S ribosomal subunit
Gram-positive and gram-negative bacteria, and several intracellular bacterial pathogens (e.g., chlamydia)
Trimethoprim
Interferes with folic acid pathway by binding the enzyme dihydrofolate reductase
Gram-positive and many gram-negative bacteria
Inhibitors of Cell Wall Synthesis
β-Lactam (Beta-Lactam) Antimicrobial Agents.
Glycopeptides and Lipopeptides.
Inhibitors of Protein Synthesis
Aminoglycosides and Aminocyclitols.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree