PART 15: Immune-Mediated, Inflammatory, and Rheumatologic Disorders
SECTION 1 | THE IMMUNE SYSTEM IN HEALTH AND DISEASE |
372e | Introduction to the Immune System |
DEFINITIONS
• Adaptive immune system—recently evolved system of immune responses mediated by T and B lymphocytes. Immune responses by these cells are based on specific antigen recognition by clonotypic receptors that are products of genes that rearrange during development and throughout the life of the organism. Additional cells of the adaptive immune system include various types of antigen-presenting cells.
• Antibody—B cell–produced molecules encoded by genes that re-arrange during B cell development consisting of immunoglobulin heavy and light chains that together form the central component of the B cell receptor for antigen. Antibody can exist as B cell–surface antigen-recognition molecules or as secreted molecules in plasma and other body fluids.
• Antigens—foreign or self-molecules that are recognized by the adaptive and innate immune systems resulting in immune cell triggering, T cell activation, and/or B cell antibody production.
• Antimicrobial peptides—small peptides <100 amino acids in length that are produced by cells of the innate immune system and have anti-infectious agent activity.
• Apoptosis—the process of programmed cell death whereby signaling through various “death receptors” on the surface of cells (e.g., tumor necrosis factor [TNF] receptors, CD95) leads to a signaling cascade that involves activation of the caspase family of molecules and leads to DNA cleavage and cell death. Apoptosis, which does not lead to induction of inordinate inflammation, is to be contrasted with cell necrosis, which does lead to induction of inflammatory responses.
• Autoimmune diseases—diseases such as systemic lupus erythematosus and rheumatoid arthritis in which cells of the adaptive immune system such as autoreactive T and B cells become overreactive and produce self-reactive T cell and antibody responses.
• Autoinflammatory diseases—hereditary disorders such as hereditary periodic fevers (HPFs) characterized by recurrent episodes of severe inflammation and fever due to mutations in controls of the innate inflammatory response, i.e., the inflammasome (see below and Table 372e-6). Patients with HPFs also have rashes and serosal and joint inflammation, and some can have neurologic symptoms. Autoinflammatory diseases are different from autoimmune diseases in that evidence for activation of adaptive immune cells such as autoreactive B cells is not present.
• B cell receptor for antigen—complex of surface molecules that rearrange during postnatal B cell development, made up of surface immunoglobulin (Ig) and associated Ig αβ chain molecules that recognize nominal antigen via Ig heavy- and light-chain variable regions, and signal the B cell to terminally differentiate to make antigen-specific antibody.
• B lymphocytes—bone marrow-derived or bursal-equivalent lymphocytes that express surface immunoglobulin (the B cell receptor for antigen) and secrete specific antibody after interaction with antigen.
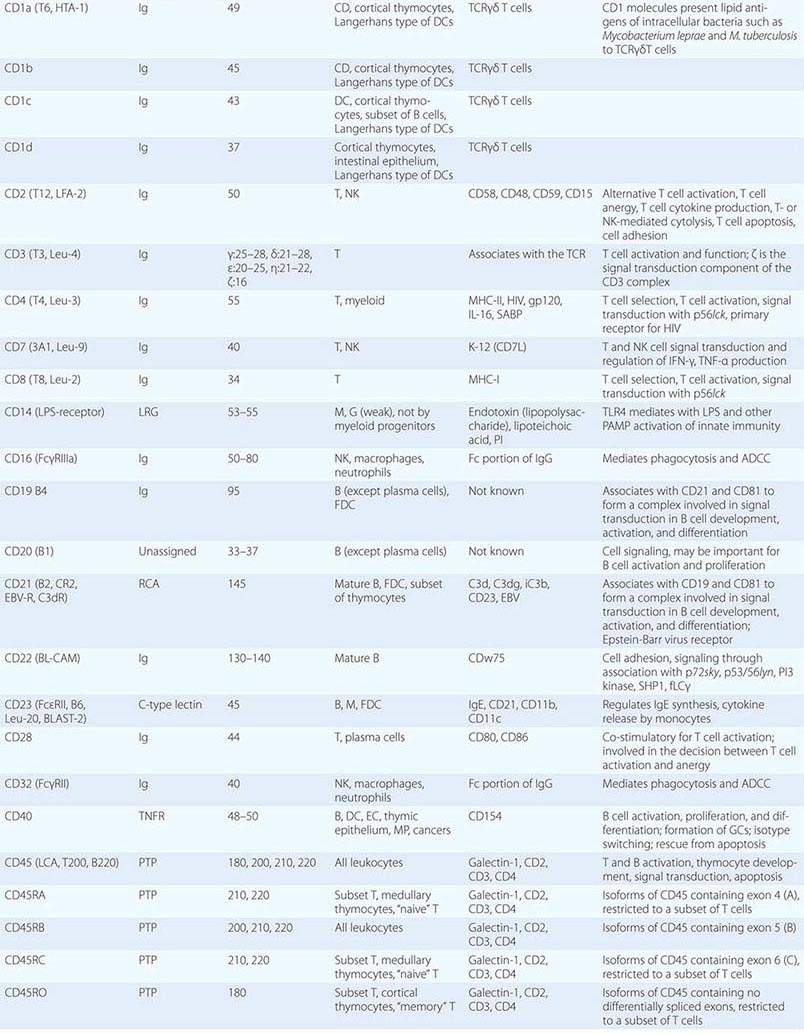
• CD classification of human lymphocyte differentiation antigens—the development of monoclonal antibody technology led to the discovery of a large number of new leukocyte surface molecules. In 1982, the First International Workshop on Leukocyte Differentiation Antigens was held to establish a nomenclature for cell-surface molecules of human leukocytes. From this and subsequent leukocyte differentiation workshops has come the cluster of differentiation (CD) classification of leukocyte antigens.
• Chemokines—soluble molecules that direct and determine immune cell movement and circulation pathways.
• Complement—cascading series of plasma enzymes and effector proteins whose function is to lyse pathogens and/or target them to be phagocytized by neutrophils and monocyte/macrophage lineage cells of the reticuloendothelial system.
• Co-stimulatory molecules—molecules of antigen-presenting cells (such as B7-1 and B7-2 or CD40) that lead to T cell activation when bound by ligands on activated T cells (such as CD28 or CD40 ligand).
• Cytokines—soluble proteins that interact with specific cellular receptors that are involved in the regulation of the growth and activation of immune cells and mediate normal and pathologic inflammatory and immune responses.
• Dendritic cells—myeloid and/or lymphoid lineage antigen-presenting cells of the adaptive immune system. Immature dendritic cells, or dendritic cell precursors, are key components of the innate immune system by responding to infections with production of high levels of cytokines. Dendritic cells are key initiators both of innate immune responses via cytokine production and of adaptive immune responses via presentation of antigen to T lymphocytes.
• Ig Fc receptors—receptors found on the surface of certain cells including B cells, natural killer cells, macrophages, neutrophils, and mast cells. Fc receptors bind to antibodies that have attached to invading pathogen-infected cells. They stimulate cytotoxic cells to destroy microbe-infected cells through a mechanism known as antibody-dependent cell-mediated cytotoxicity (ADCC). Examples of important Fc receptors include CD16 (FcγRIIIa), CD23 (FcεR), CD32 (FcγRII), CD64 (FcγRI), and CD89 (FcαR).
• Inflammasome—large cytoplasmic complexes of intracellular proteins that link the sensing of microbial products and cellular stress to the proteolytic activation of interleukin (IL)-1β and IL-18 inflammatory cytokines. Activation of molecules in the inflammasome is a key step in the response of the innate immune system for intracellular recognition of microbial and other danger signals in both health and pathologic states.
• Innate immune system—ancient immune recognition system of host cells bearing germline-encoded pattern recognition receptors that recognize pathogens and trigger a variety of mechanisms of pathogen elimination. Cells of the innate immune system include natural killer cell lymphocytes, monocytes/macrophages, dendritic cells, neutrophils, basophils, eosinophils, tissue mast cells, and epithelial cells.
• Large granular lymphocytes—lymphocytes of the innate immune system with azurophilic cytotoxic granules that have natural killer cell activity capable of killing foreign and host cells with few or no self-major histocompatibility complex (MHC) class I molecules.
• Natural killer (NK) cells—large granular lymphocytes that kill target cells expressing few or no human leukocyte antigen (HLA) class I molecules, such as malignantly transformed cells and virally infected cells. NK cells express receptors that inhibit killer cell function when self-MHC class I is present.
• NK T cells—innate-like lymphocytes that use an invariant T cell receptor (TCR)-α chain combined with a limited set of TCR-β chains and coexpress receptors commonly found on NK cells. NK T cells recognize lipid antigens of bacterial, viral, fungal, and protozoal infectious agents.
• Pathogen-associated molecular patterns (PAMPs)—Invariant molecular structures expressed by large groups of microorganisms that are recognized by host cellular pattern recognition receptors in the mediation of innate immunity.
• Pattern recognition receptors (PRRs)—germline-encoded receptors expressed by cells of the innate immune system that recognize PAMPs.
• Polyreactive natural antibodies—preexisting low-affinity antibodies produced by B cells that cross-react with multiple antigens and are available at the time of infection to bind to and coat the invading pathogen and harness innate responses to slow the infection until an adaptive high-affinity protective antibody response can be made.
• T cell exhaustion—state of T cells when the persistence of antigen disrupts memory T cell function, resulting in defects in memory T cell responses. Most frequently occurs in malignancies and in chronic viral infections such as HIV-1 and hepatitis C.
• T cell receptor (TCR) for antigen—complex of surface molecules that rearrange during postnatal T cell development made up of clonotypic TCR-α and -β chains that are associated with the CD3 complex composed of invariant γ, δ, ε, ζ, and η chains. TCR-α and -β chains recognize peptide fragments of protein antigen physically bound in antigen-presenting cell MHC class I or II molecules, leading to signaling via the CD3 complex to mediate effector functions.
• T follicular helper T cells (Tfh)—CD4 T cells in B cell follicle germinal centers that produce IL-4 and IL-21 and drive B cell development and affinity maturation in peripheral lymphoid tissues such as lymph node and spleen.
• TH17 T cells—CD4 T cells that secrete IL-17, IL-22, and IL-26 and play roles in autoimmune inflammatory disorders as well as defend against bacterial and fungal pathogens.
• T lymphocytes—thymus-derived lymphocytes that mediate adaptive cellular immune responses including T helper, T regulatory, and cytotoxic T lymphocyte effector cell functions.
• Tolerance—B and T cell nonresponsiveness to antigens that results from encounter with foreign or self-antigens by B and T lymphocytes in the absence of expression of antigen-presenting cell co-stimulatory molecules. Tolerance to antigens may be induced and maintained by multiple mechanisms either centrally (in the thymus for T cells or bone marrow for B cells) or peripherally at sites throughout the peripheral immune system.
INTRODUCTION
The human immune system has evolved over millions of years from both invertebrate and vertebrate organisms to develop sophisticated defense mechanisms to protect the host from microbes and their virulence factors. The normal immune system has three key properties: a highly diverse repertoire of antigen receptors that enables recognition of a nearly infinite range of pathogens; immune memory, to mount rapid recall immune responses; and immunologic tolerance, to avoid immune damage to normal self-tissues. From invertebrates, humans have inherited the innate immune system, an ancient defense system that uses germline-encoded proteins to recognize pathogens. Cells of the innate immune system, such as macrophages, dendritic cells, and NK lymphocytes, recognize PAMPs that are highly conserved among many microbes and use a diverse set of PRR molecules. Important components of the recognition of microbes by the innate immune system include recognition by germline-encoded host molecules, recognition of key microbe virulence factors but not recognition of self-molecules, and nonrecognition of benign foreign molecules or microbes. Upon contact with pathogens, macrophages and NK cells may kill pathogens directly or, in concert with dendritic cells, may activate a series of events that both slow the infection and recruit the more recently evolved arm of the human immune system, the adaptive immune system.
Adaptive immunity is found only in vertebrates and is based on the generation of antigen receptors on T and B lymphocytes by gene rearrangements, such that individual T or B cells express unique antigen receptors on their surface capable of specifically recognizing diverse antigens of the myriad infectious agents in the environment. Coupled with finely tuned specific recognition mechanisms that maintain tolerance (nonreactivity) to self-antigens, T and B lymphocytes bring both specificity and immune memory to vertebrate host defenses.
This chapter describes the cellular components, key molecules (Table 372e-1), and mechanisms that make up the innate and adaptive immune systems and describes how adaptive immunity is recruited to the defense of the host by innate immune responses. An appreciation of the cellular and molecular bases of innate and adaptive immune responses is critical to understanding the pathogenesis of inflammatory, autoimmune, infectious, and immunodeficiency diseases.
HUMAN LEUKOCYTE SURFACE ANTIGENS—THE CD CLASSIFICATION OF LEUKOCYTE DIFFERENTIATION ANTIGENS |
THE INNATE IMMUNE SYSTEM
All multicellular organisms, including humans, have developed the use of a limited number of surface and intracellular germline-encoded molecules that recognize large groups of pathogens. Because of the myriad human pathogens, host molecules of the human innate immune system sense “danger signals” and either recognize PAMPs, the common molecular structures shared by many pathogens, or recognize host cell molecules produced in response to infection such as heat shock proteins and fragments of the extracellular matrix. PAMPs must be conserved structures vital to pathogen virulence and survival, such as bacterial endotoxin, so that pathogens cannot mutate molecules of PAMPs to evade human innate immune responses. PRRs are host proteins of the innate immune system that recognize PAMPs as host danger signal molecules (Tables 372e-2 and 372e-3). Thus, recognition of pathogen molecules by hematopoietic and nonhematopoietic cell types leads to activation/production of the complement cascade, cytokines, and antimicrobial peptides as effector molecules. In addition, pathogen PAMPs as host danger signal molecules activate dendritic cells to mature and to express molecules on the dendritic cell surface that optimize antigen presentation to respond to foreign antigens.
MAJOR COMPONENTS OF THE INNATE IMMUNE SYSTEM |
PATTERN RECOGNITION
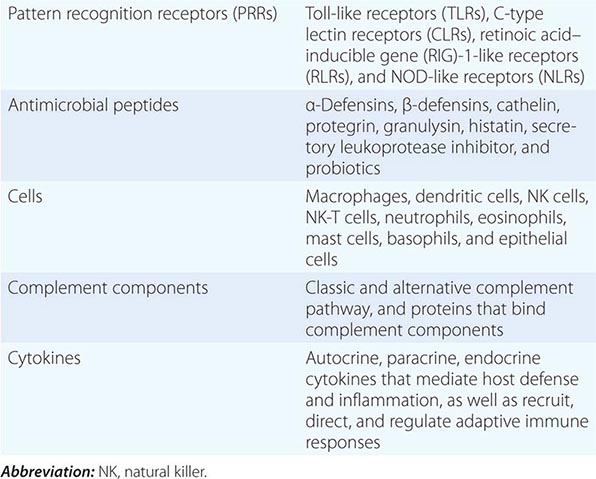
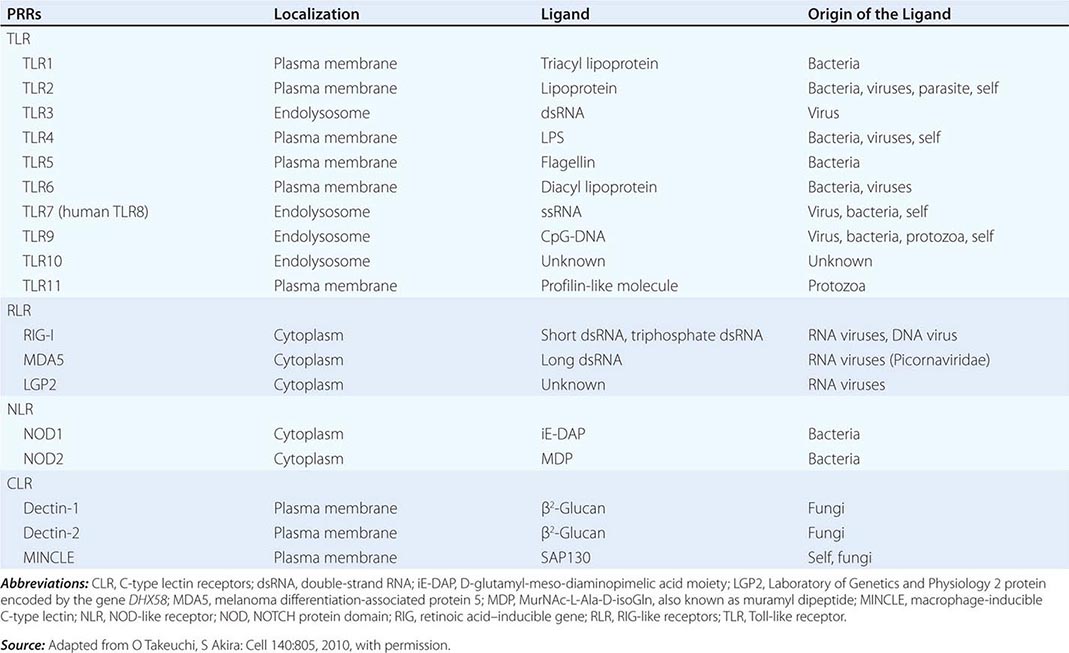
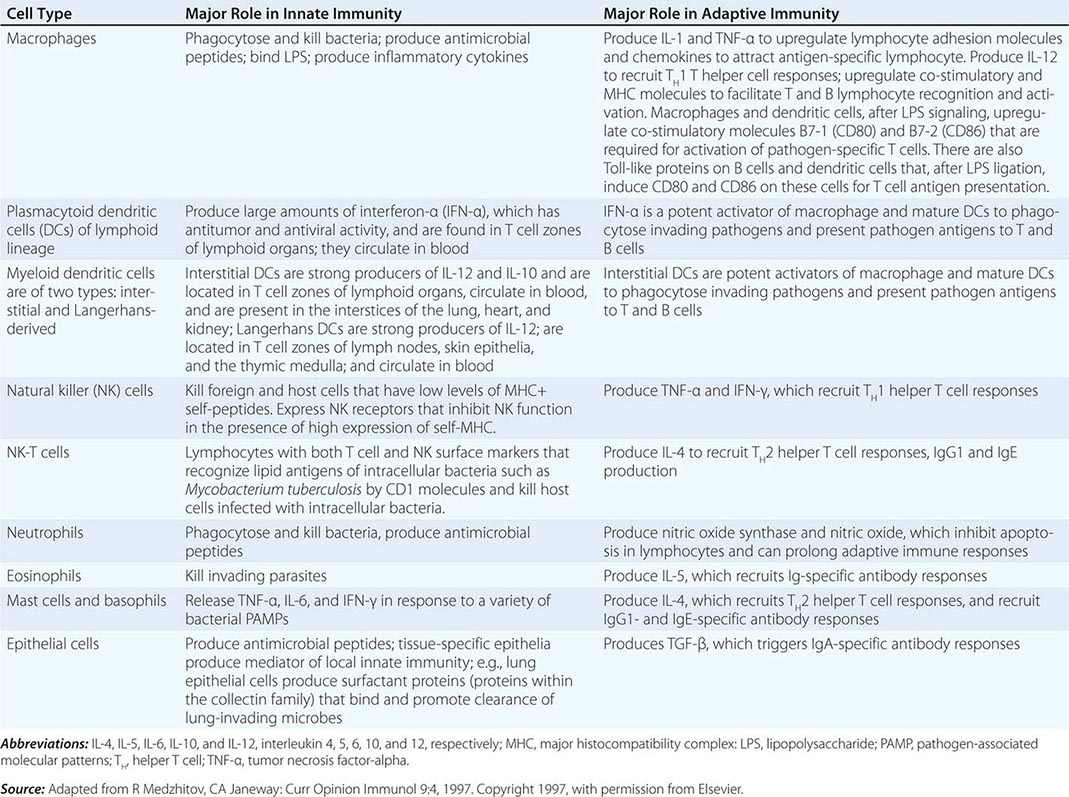
Major PRR families of proteins include transmembrane proteins, such as the Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic proteins, such as the retinoic acid–inducible gene (RIG)-1-like receptors (RLRs) and NOD-like receptors (NLRs) (Table 372e-3). A major group of PRR collagenous glycoproteins with C-type lectin domains are termed collectins and include the serum protein mannose-binding lectin (MBL). MBL and other collectins, as well as two other protein families—the pentraxins (such as C-reactive protein and serum amyloid P) and macrophage scavenger receptors—all have the property of opsonizing (coating) bacteria for phagocytosis by macrophages and can also activate the complement cascade to lyse bacteria. Integrins are cell-surface adhesion molecules that affect attachment between cells and the extracellular matrix and mediate signal transduction that reflects the chemical composition of the cell environment. For example, integrins signal after cells bind bacterial lipopolysaccharide (LPS) and activate phagocytic cells to ingest pathogens.
PATTERN RECOGNITION RECEPTORS (PRRs) AND THEIR LIGANDS |
There are multiple connections between the innate and adaptive immune systems; these include (1) a plasma protein, LPS-binding protein, that binds and transfers LPS to the macrophage LPS receptor, CD14; (2) a human family of proteins called Toll-like receptor proteins (TLRs), some of which are associated with CD14, bind LPS, and signal epithelial cells, dendritic cells, and macrophages to produce cytokines and upregulate cell-surface molecules that signal the initiation of adaptive immune responses (Fig. 372e-1, Tables 372e-3 and 372e-4), and (3) families of intracellular microbial sensors called NLRs and RLRs. Proteins in the Toll family can be expressed on macrophages, dendritic cells, and B cells as well as on a variety of nonhematopoietic cell types, including respiratory epithelial cells. Eleven TLRs have been identified in humans, and 13 TLRs have been identified in mice (Tables 372e-4 and 372e-5). Upon ligation, TLRs activate a series of intracellular events that lead to the killing of bacteria- and viral-infected cells as well as to the recruitment and ultimate activation of antigen-specific T and B lymphocytes (Fig. 372e-1). Importantly, signaling by massive amounts of LPS through TLR4 leads to the release of large amounts of cytokines that mediate LPS-induced shock. Mutations in TLR4 proteins in mice protect from LPS shock, and TLR mutations in humans protect from LPS-induced inflammatory diseases such as LPS-induced asthma (Fig. 372e-1).
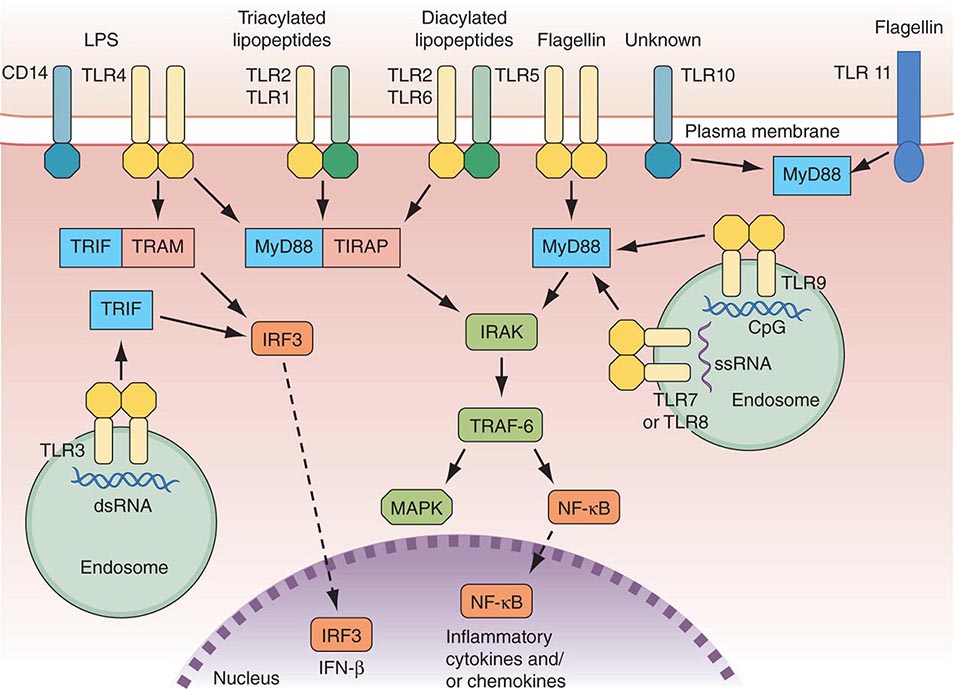
FIGURE 372e-1 Overview of major TLR signaling pathways. All TLRs signal through MyD88, with the exception of TLR3. TLR4 and the TLR2 subfamily (TLR1, TLR2, TLR6) also engage TIRAP. TLR3 signals through TRIF. TRIF is also used in conjunction with TRAM in the TLR4-MyD88-independent pathway. Dashed arrows indicate translocation into the nucleus. dsRNA, double-strand RNA; IFN, interferon; IRF3, interferon regulatory factor 3; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinases; NF-κB, nuclear factor-κB; ssRNA, single-strand RNA; TLR, Toll-like receptor. (Adapted from D van Duin et al: Trends Immunol 27:49, 2006, with permission.)
THE ROLE OF PATTERN RECOGNITION RECEPTORS (PRRs) IN MODULATION OF ADAPTIVE IMMUNE RESPONSES |
CELLS OF THE INNATE IMMUNE SYSTEM AND THEIR MAJOR ROLES IN TRIGGERING ADAPTIVE IMMUNITY |
Two other families of cytoplasmic PRRs are the NLRs and the RLRs. These families, unlike the TLRs, are composed primarily of soluble intracellular proteins that scan host cell cytoplasm for intracellular pathogens (Tables 372e-2 and 372e-3).
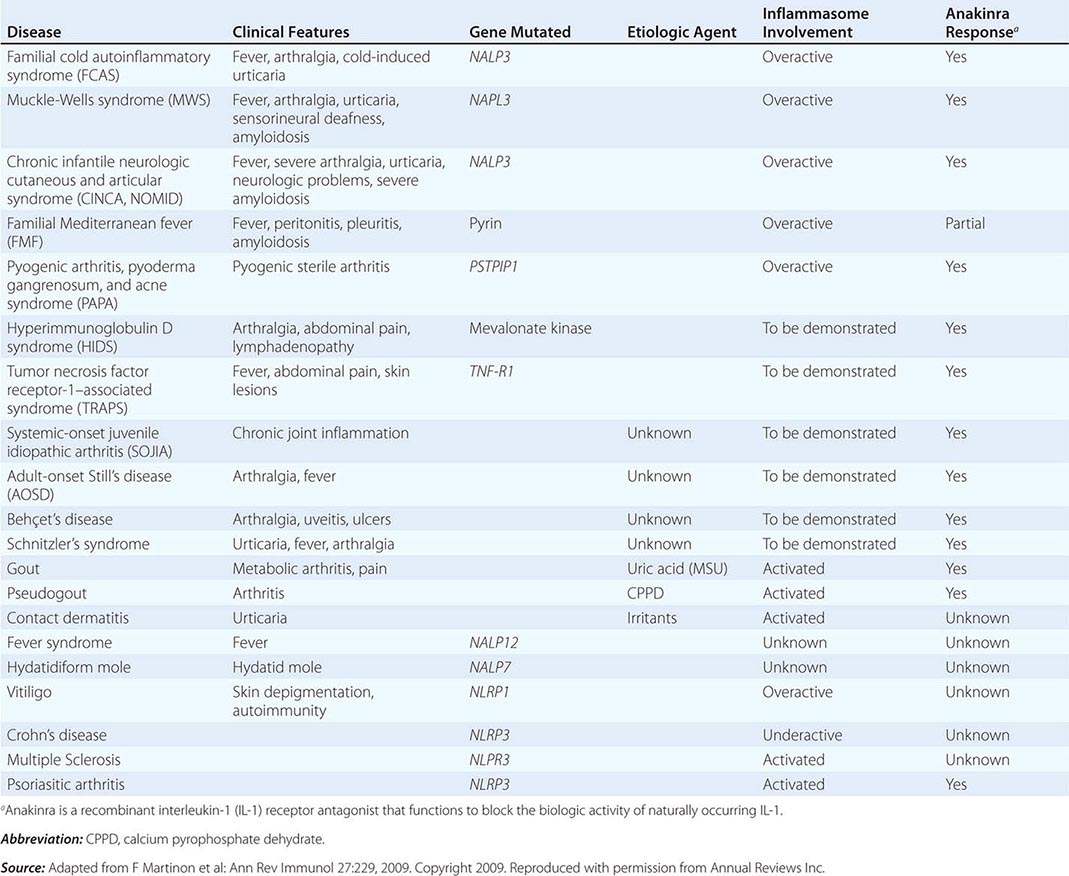
The intracellular microbial sensors, NLRs, after triggering, form large cytoplasmic complexes termed inflammasomes, which are aggregates of molecules including NOD-like receptor pyrin (NLRP) proteins that are members of the NLR family (Table 372e-3). Inflammasomes activate inflammatory caspases and IL-1β in the presence of nonbacterial danger signals (cell stress) and bacterial PAMPs. Mutations in inflammasome proteins can lead to chronic inflammation in a group of periodic febrile diseases called autoinflammatory syndromes (Table 372e-6).
DISEASES ASSOCIATED WITH INFLAMMASOME ACTIVITY |
EFFECTOR CELLS OF INNATE IMMUNITY
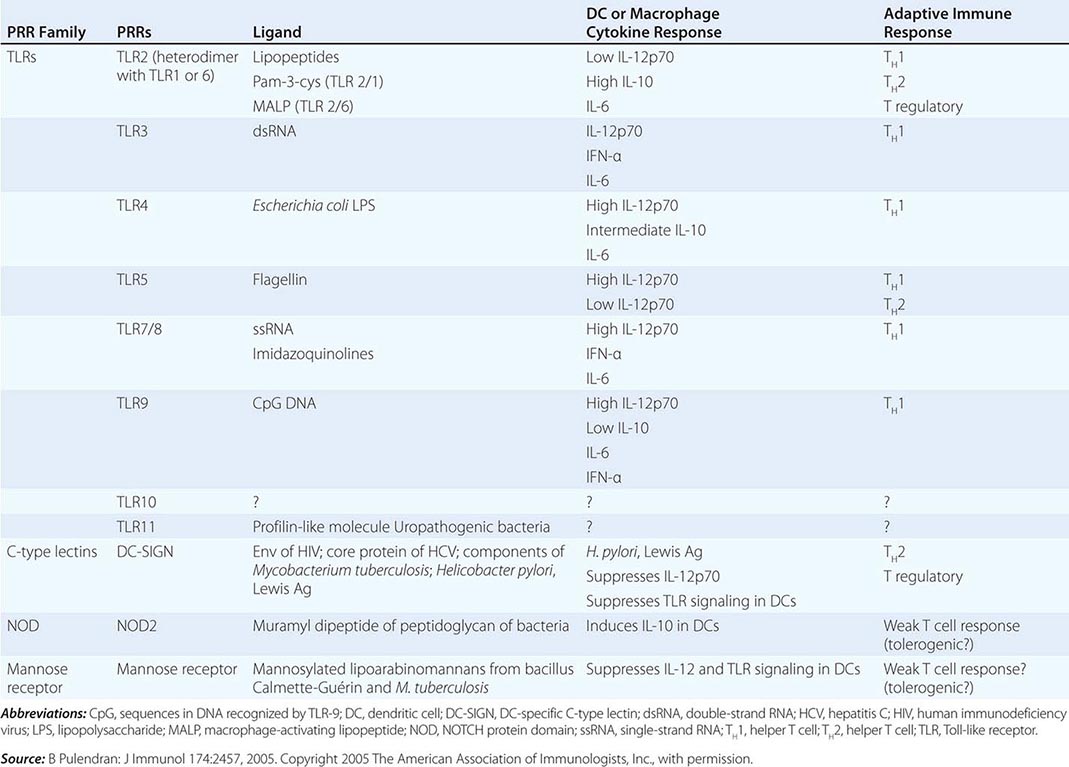
Cells of the innate immune system and their roles in the first line of host defense are listed in Table 372e-5. Equally important as their roles in the mediation of innate immune responses are the roles that each cell type plays in recruiting T and B lymphocytes of the adaptive immune system to engage in specific pathogen responses.
Monocytes-Macrophages Monocytes arise from precursor cells within bone marrow (Fig. 372e-2) and circulate with a half-life ranging from 1 to 3 days. Monocytes leave the peripheral circulation via capillaries and migration into a vast extravascular cellular pool. Tissue macrophages arise from monocytes that have migrated out of the circulation and by in situ proliferation of macrophage precursors in tissue. Common locations where tissue macrophages (and certain of their specialized forms) are found are lymph node, spleen, bone marrow, perivascular connective tissue, serous cavities such as the peritoneum, pleura, skin connective tissue, lung (alveolar macrophages), liver (Kupffer cells), bone (osteoclasts), central nervous system (microglia cells), and synovium (type A lining cells).
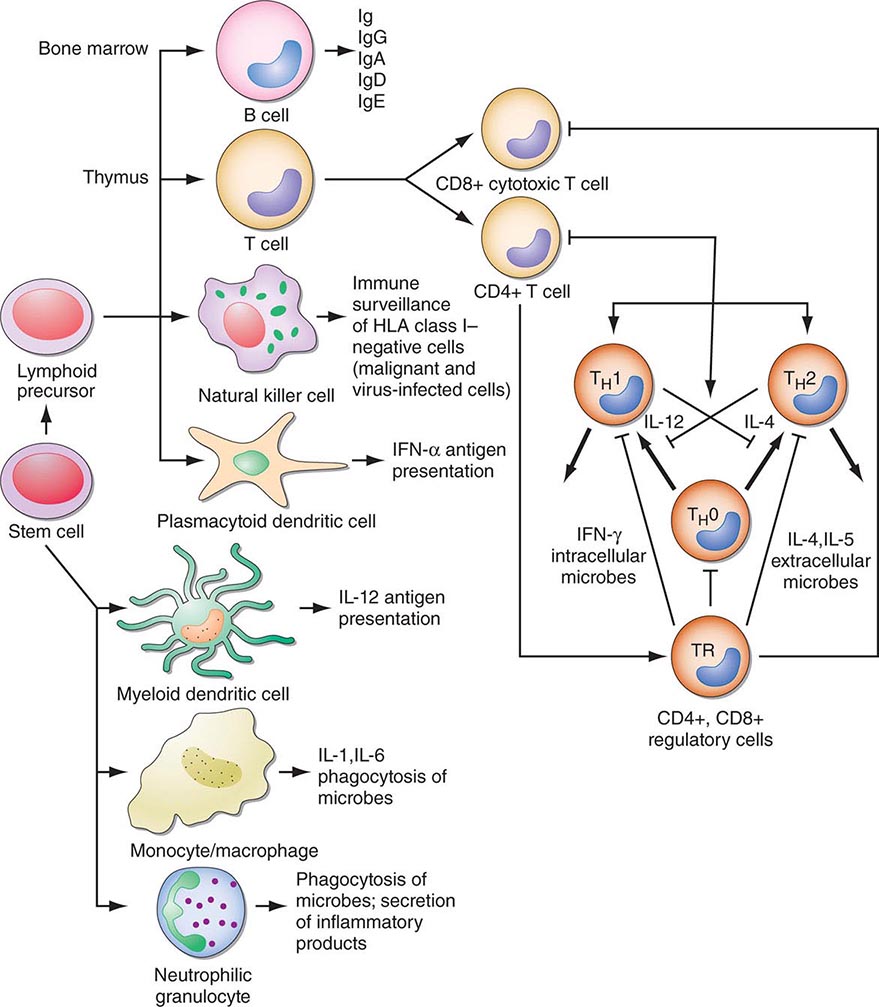
FIGURE 372e-2 Schematic model of intercellular interactions of adaptive immune system cells. In this figure, the arrows denote that cells develop from precursor cells or produce cytokines or antibodies; lines ending with bars indicate suppressive intercellular interactions. Stem cells differentiate into either T cells, antigen-presenting dendritic cells, natural killer cells, macrophages, granulocytes, or B cells. Foreign antigen is processed by dendritic cells, and peptide fragments of foreign antigen are presented to CD4+ and/or CD8+ T cells. CD8+ T cell activation leads to induction of cytotoxic T lymphocyte (CTL) or killer T cell generation, as well as induction of cytokine-producing CD8+ cytotoxic T cells. For antibody production against the same antigen, active antigen is bound to sIg within the B cell receptor complex and drives B cell maturation into plasma cells that secrete Ig. TH1 or TH2 CD4+ T cells producing interleukin (IL) 4, IL-5, or interferon (IFN) γ regulate the Ig class switching and determine the type of antibody produced. TH17 cells secrete IL-17, IL-22, IL-26, which contribute to host defense against extracellular bacteria and fungi, particularly at mucosal surfaces. CD4+, CD25+ T regulatory cells produce IL-10 and downregulate T and B cell responses once the microbe has been eliminated. GM-CSF, granulocyte-macrophage colony-stimulating factor; TNF, tumor necrosis factor.
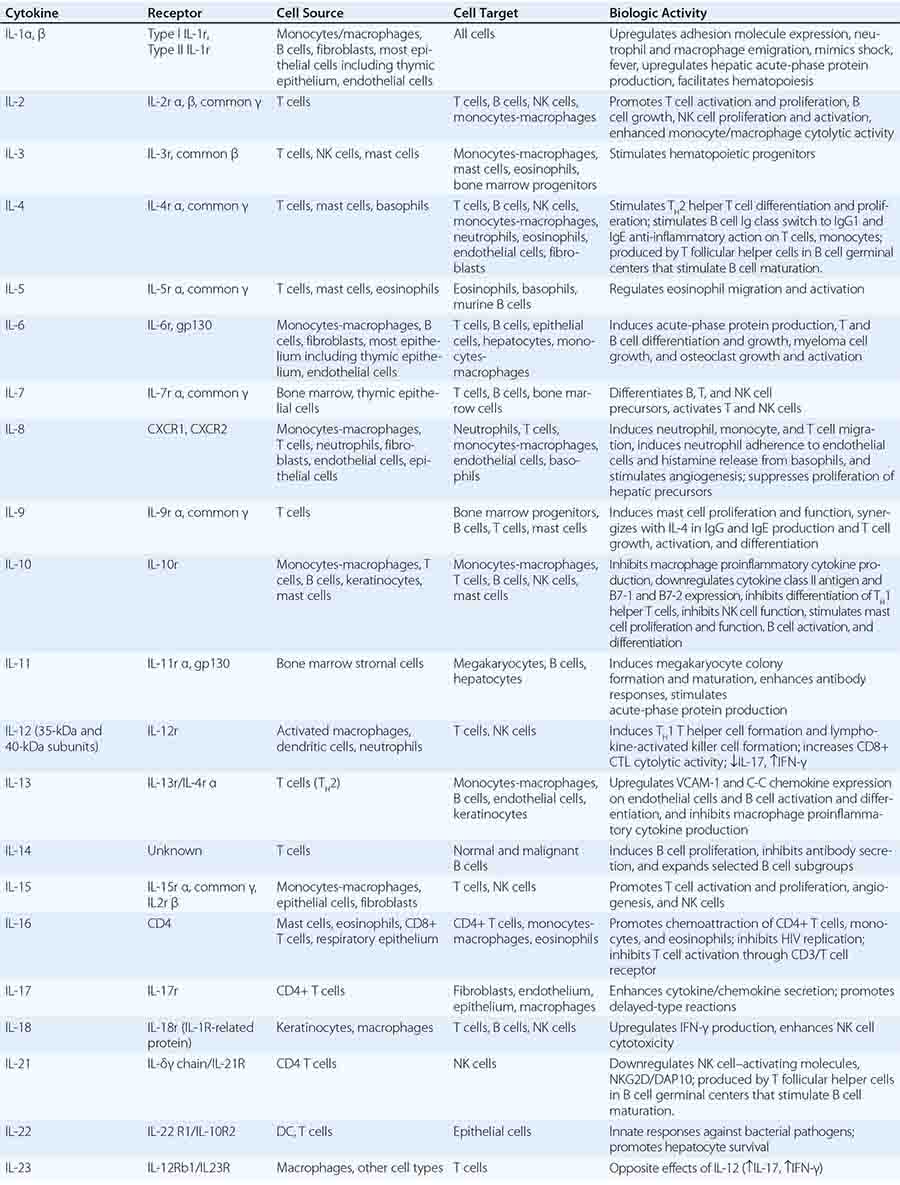
In general, monocytes-macrophages are on the first line of defense associated with innate immunity and ingest and destroy microorganisms through the release of toxic products such as hydrogen peroxide (H2O2) and nitric oxide (NO). Inflammatory mediators produced by macrophages attract additional effector cells such as neutrophils to the site of infection. Macrophage mediators include prostaglandins; leukotrienes; platelet activating factor; cytokines such as IL-1, TNF-α, IL-6, and IL-12; and chemokines (Tables 372e-7 to 372e-9).
CYTOKINES AND CYTOKINE RECEPTORS |
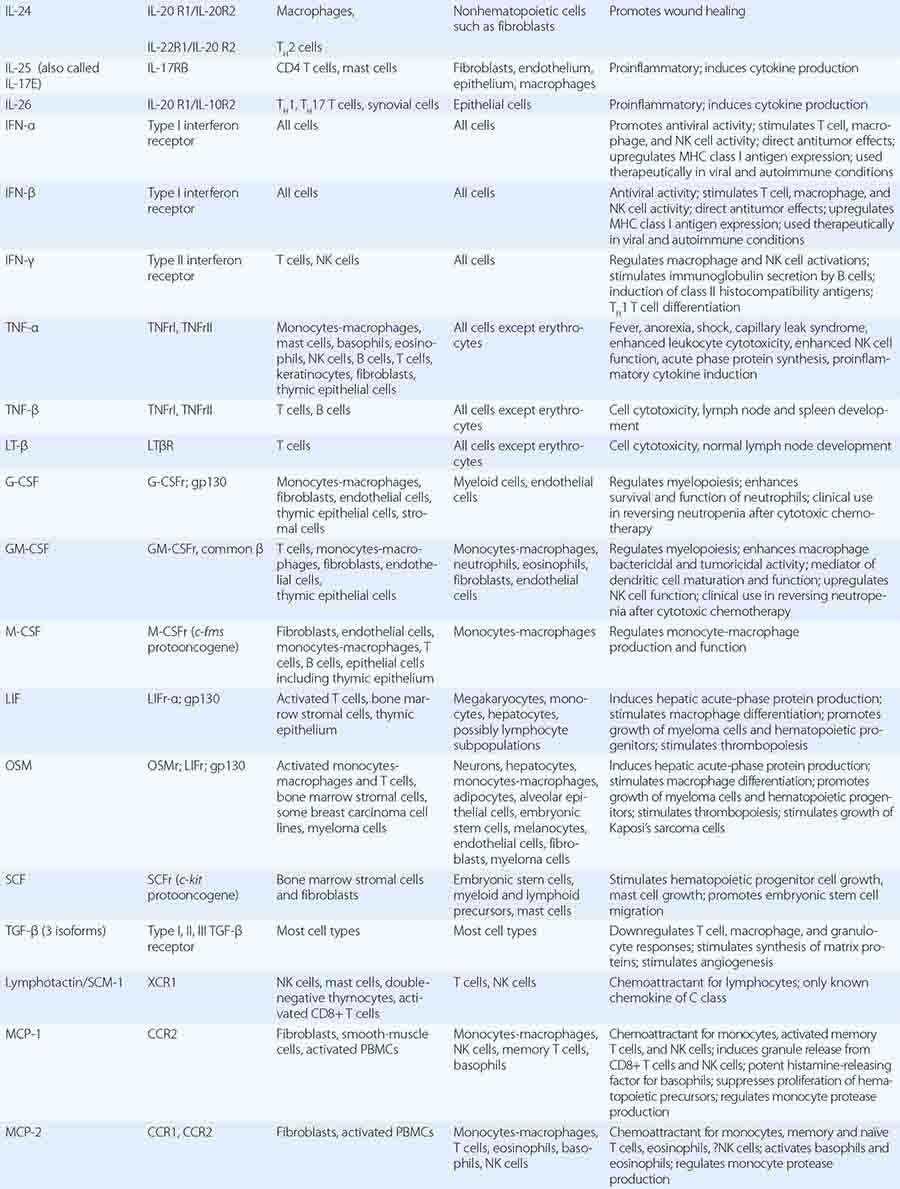
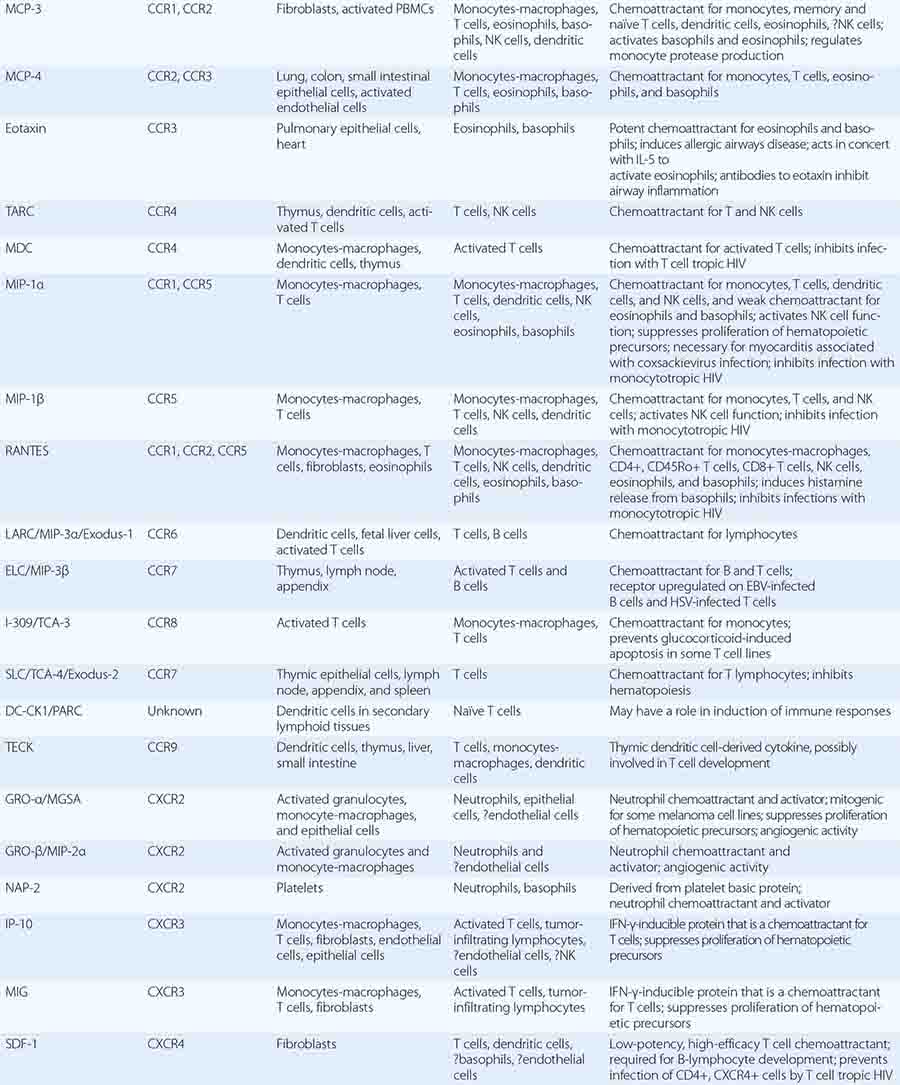

CC, CXC1, CX3, C1, AND XC FAMILIES OF CHEMOKINES AND CHEMOKINE RECEPTORS |
CYTOKINE FAMILIES GROUPED BY STRUCTURAL SIMILARITY |
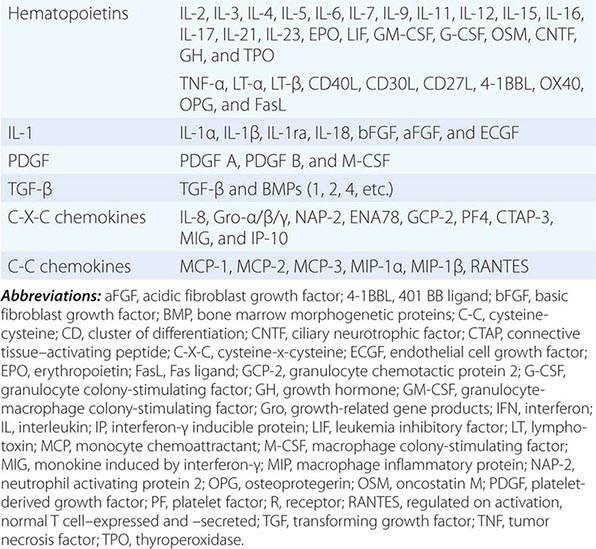
Although monocytes-macrophages were originally thought to be the major antigen-presenting cells (APCs) of the immune system, it is now clear that cell types called dendritic cells are the most potent and effective APCs in the body (see below). Monocytes-macrophages mediate innate immune effector functions such as destruction of antibody-coated bacteria, tumor cells, or even normal hematopoietic cells in certain types of autoimmune cytopenias. Monocytes-macrophages ingest bacteria or are infected by viruses, and in doing so, they frequently undergo programmed cell death or apoptosis. Macrophages that are infected by intracellular infectious agents are recognized by dendritic cells as infected and apoptotic cells and are phagocytosed by dendritic cells. In this manner, dendritic cells “cross-present” infectious agent antigens of macrophages to T cells. Activated macrophages can also mediate antigen-nonspecific lytic activity and eliminate cell types such as tumor cells in the absence of antibody. This activity is largely mediated by cytokines (i.e., TNF-α and IL-1). Monocytes-macrophages express lineage-specific molecules (e.g., the cell-surface LPS receptor, CD14) as well as surface receptors for a number of molecules, including the Fc region of IgG, activated complement components, and various cytokines (Table 372e-7).
Dendritic Cells Human dendritic cells (DCs) contain several subsets, including myeloid DCs and plasmacytoid DCs. Myeloid DCs can differentiate into either macrophages-monocytes or tissue-specific DCs. In contrast to myeloid DCs, plasmacytoid DCs are inefficient APCs but are potent producers of type I interferon (IFN) (e.g., IFN-α) in response to viral infections. The maturation of DCs is regulated through cell-to-cell contact and soluble factors, and DCs attract immune effectors through secretion of chemokines. When DCs come in contact with bacterial products, viral proteins, or host proteins released as danger signals from distressed host cells (Figs. 372e-2 and 372e-3), infectious agent molecules bind to various TLRs and activate DCs to release cytokines and chemokines that drive cells of the innate immune system to become activated to respond to the invading organism, and recruit T and B cells of the adaptive immune system to respond. Plasmacytoid DCs produce antiviral IFN-α that activates NK cell killing of pathogen-infected cells; IFN-α also activates T cells to mature into antipathogen cytotoxic (killer) T cells. Following contact with pathogens, both plasmacytoid and myeloid DCs produce chemokines that attract helper and cytotoxic T cells, B cells, polymorphonuclear cells, and naïve and memory T cells as well as regulatory T cells to ultimately dampen the immune response once the pathogen is controlled. TLR engagement on DCs upregulates MHC class II, B7-1 (CD80), and B7-2 (CD86), which enhance DC-specific antigen presentation and induce cytokine production (Table 372e-7). Thus, DCs are important bridges between early (innate) and later (adaptive) immunity. DCs also modulate and determine the types of immune responses induced by pathogens via the TLRs expressed on DCs (TLR7–9 on plasmacytoid DCs, TLR4 on monocytoid DCs) and via the TLR adapter proteins that are induced to associate with TLRs (Fig. 372e-1, Table 372e-4). In addition, other PRRs, such as C-type lectins, NLRs, and mannose receptors, upon ligation by pathogen products, activate cells of the adaptive immune system and, like TLR stimulation, by a variety of factors, determine the type and quality of the adaptive immune response that is triggered (Table 372e-4).
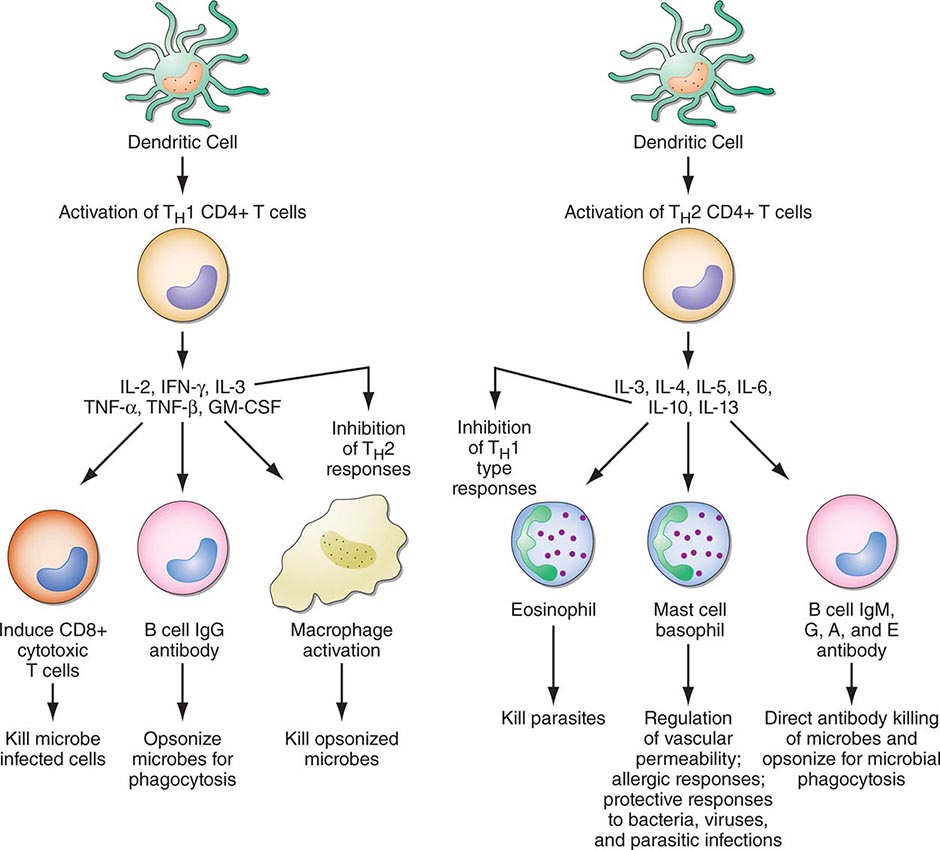
FIGURE 372e-3 CD4+ helper T1 (TH1) cells and TH2 T cells secrete distinct but overlapping sets of cytokines. TH1 CD4+ cells are frequently activated in immune and inflammatory reactions against intracellular bacteria or viruses, whereas TH2 CD4+ cells are frequently activated for certain types of antibody production against parasites and extracellular encapsulated bacteria; they are also activated in allergic diseases. GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor. (Adapted from S Romagnani: CD4 effector cells, in Inflammation: Basic Principles and Clinical Correlates, 3rd ed, J Gallin, R Snyderman [eds]. Philadelphia, Lippincott Williams & Wilkins, 1999, p 177; with permission.)
Large Granular Lymphocytes/Natural Killer Cells Large granular lymphocytes (LGLs) or NK cells account for ~5–15% of peripheral blood lymphocytes. NK cells are nonadherent, nonphagocytic cells with large azurophilic cytoplasmic granules. NK cells express surface receptors for the Fc portion of IgG (FcR) (CD16) and for NCAM-I (CD56), and many NK cells express T lineage markers, particularly CD8, and proliferate in response to IL-2. NK cells arise in both bone marrow and thymic microenvironments.
Functionally, NK cells share features with both monocytes-macrophages and neutrophils in that they mediate both ADCC and NK cell activity. ADCC is the binding of an opsonized (antibody-coated) target cell to an Fc receptor-bearing effector cell via the Fc region of antibody, resulting in lysis of the target by the effector cell. NK cell cytotoxicity is the nonimmune (i.e., effector cell never having had previous contact with the target), MHC-unrestricted, non-antibody-mediated killing of target cells, which are usually malignant cell types, transplanted foreign cells, or virus-infected cells. Thus, NK cell cytotoxicity may play an important role in immune surveillance and destruction of malignant and virus-infected host cells. NK cell hyporesponsiveness is also observed in patients with Chédiak-Higashi syndrome, an autosomal recessive disease associated with fusion of cytoplasmic granules and defective degranulation of neutrophil lysosomes.
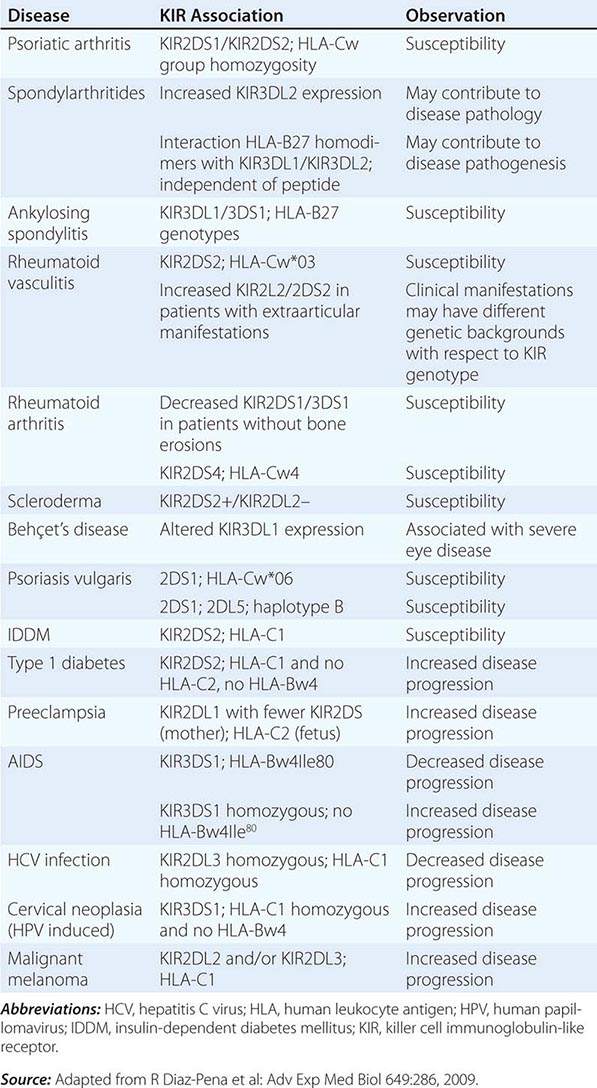
NK cells have a variety of surface receptors that have inhibitory or activating functions and belong to two structural families. These families include the immunoglobulin superfamily and the lectin-like type II transmembrane proteins. NK immunoglobulin superfamily receptors include the killer cell immunoglobulin-like activating or inhibitory receptors (KIRs), many of which have been shown to have HLA class I ligands. The KIRs are made up proteins with either two (KIR2D) or three (KIR3D) extracellular immunoglobulin domains (D). Moreover, their nomenclature designates their function as either inhibitory KIRs with a long (L) cytoplasmic tail and immunoreceptor tyrosine-based inhibitory motif (ITIM) (KIRDL) or activating KIRs with a short (S) cytoplasmic tail (KIRDS). NK cell inactivation by KIRs is a central mechanism to prevent damage to normal host cells. Genetic studies have demonstrated the association of KIRs with viral infection outcome and autoimmune disease (Table 372e-10).
ASSOCIATION OF KIRs WITH DISEASE |
In addition to the KIRs, a second set of immunoglobulin superfamily receptors includes the natural cytotoxicity receptors (NCRs), which include NKp46, NKp30, and NKp44. These receptors help to mediate NK cell activation against target cells. The ligands to which NCRs bind on target cells have been recently recognized to be comprised of molecules of pathogens such as influenza, vaccinia, and malaria as well as host molecules expressed on tumor cells.
NK cell signaling is, therefore, a highly coordinated series of inhibiting and activating signals that prevent NK cells from responding to uninfected, nonmalignant self-cells; however, they are activated to attack malignant and virally infected cells (Fig. 372e-4). Recent evidence suggests that NK cells, although not possessing rearranging immune recognition genes, may be able to mediate recall for NK cell responses to viruses and for immune responses such as contact hypersensitivity.
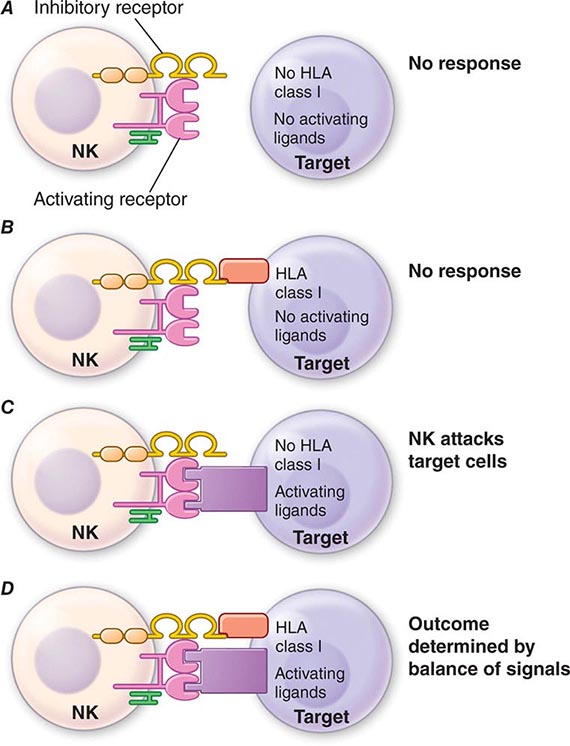
FIGURE 372e-4 Encounters between NK cells: potential targets and possible outcomes. The amount of activating and inhibitory receptors on the NK cells and the amount of ligands on the target cell, as well as the qualitative differences in the signals transduced, determine the extent of the NK response. A. When target cells have no HLA class I or activating ligands, NK cells cannot kill target cells. B. When target cells bear self-HLA, NK cells cannot kill targets. C. When target cells are pathogen infected and have downregulated HLA and express activating ligands, NK cells kill target cells. D. When NK cells encounter targets with both self-HLA and activating receptors, then the level of target killing is determined by the balance of inhibitory and activating signals to the NK cell. HLA, human leukocyte antigen; NK, natural killer. (Adapted from L Lanier: Annu Rev Immunol 23:225, 2005; reproduced with permission from Annual Reviews Inc. Copyright 2011 by Annual Reviews Inc.)
Some NK cells express CD3 and invariant TCR-α chains and are termed NK T cells. TCRs of NK T cells recognize lipid molecules of intracellular bacteria when presented in the context of CD1d molecules on APCs. Upon activation, NK T cells secrete effector cytokines such as IL-4 and IFN-γ. This mode of recognition of intracellular bacteria such as Listeria monocytogenes and Mycobacterium tuberculosis by NK T cells leads to induction of activation of DCs and is thought to be an important innate defense mechanism against these organisms.
The receptors for the Fc portion of IgG (FcγRs) are present on NK cells, B cells, macrophages, neutrophils, and mast cells and mediate interactions of IgG with antibody-coated target cells, such as virally infected cells. Antibody-NK interaction via antibody Fc and NK cell FcR links the adaptive and innate immune systems and regulates the mediation of IgG antibody effector functions such as ADCC. There are both activation and inhibitory FcγRs. Activation FcRs, such as FcγRI (CD64), FcγRII (CD32), and FcγRII (CD64), are characterized by the presence of an immunoreceptor tyrosine-based activating motif (ITAM) sequence, whereas inhibitory FcRs, such as FcγRIIb, contain an immunoreceptor tyrosine-based inhibitory motif (ITIM) sequence. There is evidence that dysregulation in IgG-FcγR interactions plays a role in arthritis, multiple sclerosis, and systemic lupus erythematosus.
Neutrophils, Eosinophils, and Basophils Granulocytes are present in nearly all forms of inflammation and are amplifiers and effectors of innate immune responses (Figs. 372e-2 and 372e-3). Unchecked accumulation and activation of granulocytes can lead to host tissue damage, as seen in neutrophil- and eosinophil-mediated systemic necrotizing vasculitis. Granulocytes are derived from stem cells in bone marrow. Each type of granulocyte (neutrophil, eosinophil, or basophil) is derived from a different subclass of progenitor cell that is stimulated to proliferate by colony-stimulating factors (Table 372e-7). During terminal maturation of granulocytes, class-specific nuclear morphology and cytoplasmic granules appear that allow for histologic identification of granulocyte type.
Neutrophils express Fc receptor IIIa for IgG (CD16) as well as receptors for activated complement components (C3b or CD35). Upon interaction of neutrophils with antibody-coated (opsonized) bacteria or immune complexes, azurophilic granules (containing myeloperoxidase, lysozyme, elastase, and other enzymes) and specific granules (containing lactoferrin, lysozyme, collagenase, and other enzymes) are released, and microbicidal superoxide radicals (O2–) are generated at the neutrophil surface. The generation of superoxide leads to inflammation by direct injury to tissue and by alteration of macromolecules such as collagen and DNA.
Eosinophils express Fc receptor II for IgG (CD32) and are potent cytotoxic effector cells for various parasitic organisms. In Nippostrongylus brasiliensis helminth infection, eosinophils are important cytotoxic effector cells for removal of these parasites. Key to regulation of eosinophil cytotoxicity to N. brasiliensis worms are antigen-specific T helper cells that produce IL-4, thus providing an example of regulation of innate immune responses by adaptive immunity antigen-specific T cells. Intracytoplasmic contents of eosinophils, such as major basic protein, eosinophil cationic protein, and eosinophil-derived neurotoxin, are capable of directly damaging tissues and may be responsible in part for the organ system dysfunction in the hypereosinophilic syndromes (Chap. 80). Because the eosinophil granule contains anti-inflammatory types of enzymes (histaminase, arylsulfatase, phospholipase D), eosinophils may homeostatically downregulate or terminate ongoing inflammatory responses.
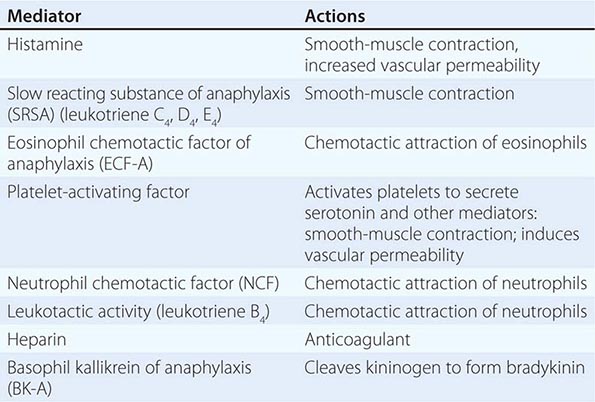
Basophils and tissue mast cells are potent reservoirs of cytokines such as IL-4 and can respond to bacteria and viruses with antipathogen cytokine production through multiple TLRs expressed on their surface. Mast cells and basophils can also mediate immunity through the binding of antipathogen antibodies. This is a particularly important host defense mechanism against parasitic diseases. Basophils express high-affinity surface receptors for IgE (FcεRII) (CD23) and, upon cross-linking of basophil-bound IgE by antigen, can release histamine, eosinophil chemotactic factor of anaphylaxis, and neutral protease—all mediators of allergic immediate (anaphylaxis) hypersensitivity responses (Table 372e-11). In addition, basophils express surface receptors for activated complement components (C3a, C5a), through which mediator release can be directly affected. Thus, basophils, like most cells of the immune system, can be activated in the service of host defense against pathogens, or they can be activated for mediation release and cause pathogenic responses in allergic and inflammatory diseases. For further discussion of tissue mast cells, see Chap. 376.
EXAMPLES OF MEDIATORS RELEASED FROM IMMUNE CELLS AND BASOPHILS |
The Complement System The complement system, an important soluble component of the innate immune system, is a series of plasma enzymes, regulatory proteins, and proteins that are activated in a cascading fashion, resulting in cell lysis. There are four pathways of the complement system: the classic activation pathway activated by antigen/antibody immune complexes, the mannose-binding lectin (MBL) (a serum collectin; Table 372e-3) activation pathway activated by microbes with terminal mannose groups, the alternative activation pathway activated by microbes or tumor cells, and the terminal pathway that is common to the first three pathways and leads to the membrane attack complex that lyses cells (Fig. 372e-5). The series of enzymes of the complement system are serine proteases.
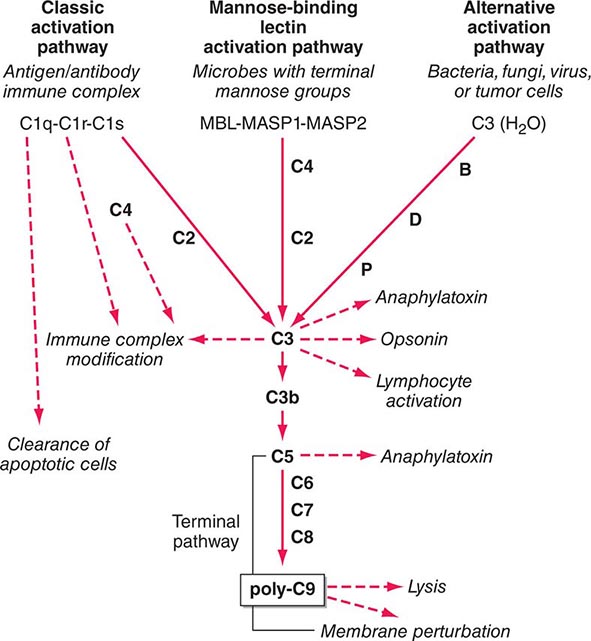
FIGURE 372e-5 The four pathways and the effector mechanisms of the complement system. Dashed arrows indicate the functions of pathway components. (After BJ Morley, MJ Walport: The Complement Facts Books. London, Academic Press, 2000, Chap. 2; with permission. Copyright Academic Press, London, 2000.)
Activation of the classic complement pathway via immune complex binding to C1q links the innate and adaptive immune systems via specific antibody in the immune complex. The alternative complement activation pathway is antibody-independent and is activated by binding of C3 directly to pathogens and “altered self” such as tumor cells. In the renal glomerular inflammatory disease IgA nephropathy, IgA activates the alternative complement pathway and causes glomerular damage and decreased renal function. Activation of the classic complement pathway via C1, C4, and C2 and activation of the alternative pathway via factor D, C3, and factor B both lead to cleavage and activation of C3. C3 activation fragments, when bound to target surfaces such as bacteria and other foreign antigens, are critical for opsonization (coating by antibody and complement) in preparation for phagocytosis. The MBL pathway substitutes MBL-associated serine proteases (MASPs) 1 and 2 for C1q, C1r, and C1s to activate C4. The MBL activation pathway is activated by mannose on the surface of bacteria and viruses.
The three pathways of complement activation all converge on the final common terminal pathway. C3 cleavage by each pathway results in activation of C5, C6, C7, C8, and C9, resulting in the membrane attack complex that physically inserts into the membranes of target cells or bacteria and lyses them.
Thus, complement activation is a critical component of innate immunity for responding to microbial infection. The functional consequences of complement activation by the three initiating pathways and the terminal pathway are shown in Fig. 372e-5. In general the cleavage products of complement components facilitate microbe or damaged cell clearance (C1q, C4, C3), promote activation and enhancement of inflammation (anaphylatoxins, C3a, C5a), and promote microbe or opsonized cell lysis (membrane attack complex).
CYTOKINES
Cytokines are soluble proteins produced by a wide variety of cell types (Tables 372e-7 to 372e-9). They are critical for both normal innate and adaptive immune responses, and their expression may be perturbed in most immune, inflammatory, and infectious disease states.
Cytokines are involved in the regulation of the growth, development, and activation of immune system cells and in the mediation of the inflammatory response. In general, cytokines are characterized by considerable redundancy; different cytokines have similar functions. In addition, many cytokines are pleiotropic in that they are capable of acting on many different cell types. This pleiotropism results from the expression on multiple cell types of receptors for the same cytokine (see below), leading to the formation of “cytokine networks.” The action of cytokines may be (1) autocrine when the target cell is the same cell that secretes the cytokine, (2) paracrine when the target cell is nearby, and (3) endocrine when the cytokine is secreted into the circulation and acts distal to the source.
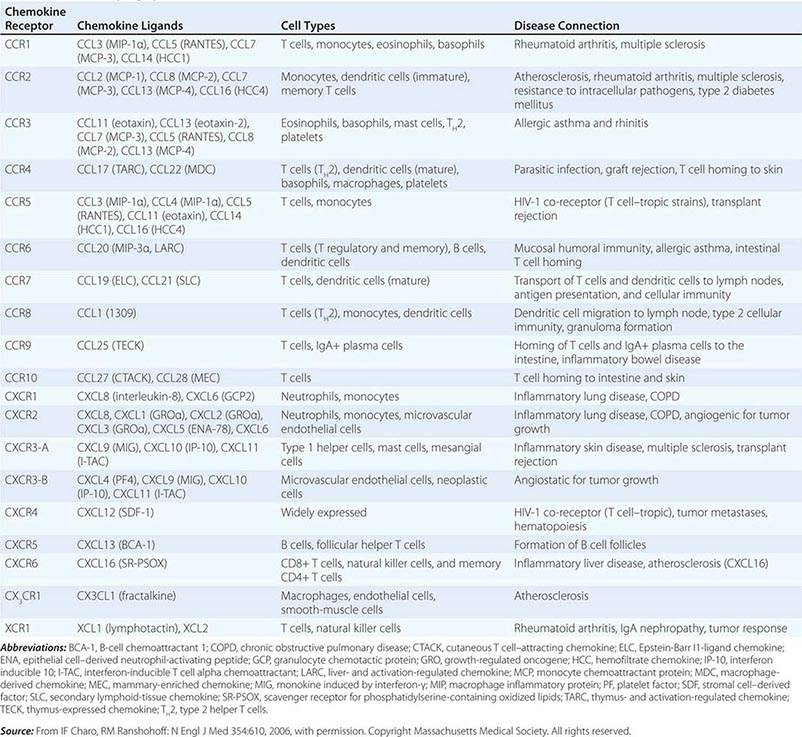
Cytokines have been named based on presumed targets or based on presumed functions. Those cytokines that are thought to primarily target leukocytes have been named interleukins (IL-1, -2, -3, etc.). Many cytokines that were originally described as having a certain function have retained those names (e.g., granulocyte colony-stimulating factor [G-CSF]). Cytokines belong in general to three major structural families: the hematopoietin family; the TNF, IL-1, platelet-derived growth factor (PDGF), and transforming growth factor (TGF) β families; and the CXC and C-C chemokine families (Table 372e-9). Chemokines are cytokines that regulate cell movement and trafficking; they act through G protein-coupled receptors and have a distinctive three-dimensional structure. IL-8 is the only chemokine that early on was named an IL (Table 372e-7).
In general, cytokines exert their effects by influencing gene activation that results in cellular activation, growth, differentiation, functional cell-surface molecule expression, and cellular effector function. In this regard, cytokines can have dramatic effects on the regulation of immune responses and the pathogenesis of a variety of diseases. Indeed, T cells have been categorized on the basis of the pattern of cytokines that they secrete, which results in either humoral immune response (TH2) or cell-mediated immune response (TH1). A third type of T helper cell is the TH17 cell that contributes to host defense against extracellular bacteria and fungi, particularly at mucosal sites (Fig. 372e-2).
Cytokine receptors can be grouped into five general families based on similarities in their extracellular amino acid sequences and conserved structural domains. The immunoglobulin (Ig) superfamily represents a large number of cell-surface and secreted proteins. The IL-1 receptors (type 1, type 2) are examples of cytokine receptors with extracellular Ig domains.
The hallmark of the hematopoietic growth factor (type 1) receptor family is that the extracellular regions of each receptor contain two conserved motifs. One motif, located at the N terminus, is rich in cysteine residues. The other motif is located at the C terminus proximal to the transmembrane region and comprises five amino acid residues, tryptophan-serine-X-tryptophan-serine (WSXWS). This family can be grouped on the basis of the number of receptor subunits they have and on the utilization of shared subunits. A number of cytokine receptors, i.e., IL-6, IL-11, IL-12, and leukemia inhibitory factor, are paired with gp130. There is also a common 150-kDa subunit shared by IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF) receptors. The gamma chain (γc) of the IL-2 receptor is common to the IL-2, IL-4, IL-7, IL-9, and IL-15 receptors. Thus, the specific cytokine receptor is responsible for ligand-specific binding, whereas the subunits such as gp130, the 150-kDa subunit, and γc are important in signal transduction. The γc gene is on the × chromosome, and mutations in the γc protein result in the X-linked form of severe combined immune deficiency syndrome (X-SCID) (Chap. 374).
The members of the interferon (type II) receptor family include the receptors for IFN-γ and -β, which share a similar 210-amino-acid binding domain with conserved cysteine pairs at both the amino and carboxy termini. The members of the TNF (type III) receptor family share a common binding domain composed of repeated cysteine-rich regions. Members of this family include the p55 and p75 receptors for TNF (TNF-R1 and TNF-R2, respectively); CD40 antigen, which is an important B cell-surface marker involved in immunoglobulin isotype switching; fas/Apo-1, whose triggering induces apoptosis; CD27 and CD30, which are found on activated T cells and B cells; and nerve growth factor receptor.
The common motif for the seven transmembrane helix family was originally found in receptors linked to GTP-binding proteins. This family includes receptors for chemokines (Table 372e-8), β-adrenergic receptors, and retinal rhodopsin. It is important to note that two members of the chemokine receptor family, CXC chemokine receptor type 4 (CXCR4) and β chemokine receptor type 5 (CCR5), have been found to serve as the two major co-receptors for binding and entry of HIV into CD4-expressing host cells (Chap. 226).
Significant advances have been made in defining the signaling pathways through which cytokines exert their intracellular effects. The Janus family of protein tyrosine kinases (JAK) is a critical element involved in signaling via the hematopoietin receptors. Four JAK kinases, JAK1, JAK2, JAK3, and Tyk2, preferentially bind different cytokine receptor subunits. Cytokine binding to its receptor brings the cytokine receptor subunits into apposition and allows a pair of JAKs to transphosphorylate and activate one another. The JAKs then phosphorylate the receptor on the tyrosine residues and allow signaling molecules to bind to the receptor, whereby the signaling molecules become phosphorylated. Signaling molecules bind the receptor because they have domains (SH2, or src homology 2 domains) that can bind phosphorylated tyrosine residues. There are a number of these important signaling molecules that bind the receptor, such as the adapter molecule SHC, which can couple the receptor to the activation of the mitogen-activated protein kinase pathway. In addition, an important class of substrate of the JAKs is the signal transducers and activators of transcription (STAT) family of transcription factors. STATs have SH2 domains that enable them to bind to phosphorylated receptors, where they are then phosphorylated by the JAKs. It appears that different STATs have specificity for different receptor subunits. The STATs then dissociate from the receptor and translocate to the nucleus, bind to DNA motifs that they recognize, and regulate gene expression. The STATs preferentially bind DNA motifs that are slightly different from one another and thereby control transcription of specific genes. The importance of this pathway is particularly relevant to lymphoid development. Mutations of JAK3 itself also result in a disorder identical to X-SCID; however, because JAK3 is found on chromosome 19 and not on the × chromosome, JAK3 deficiency occurs in boys and girls (Chap. 374).
THE ADAPTIVE IMMUNE SYSTEM
Adaptive immunity is characterized by antigen-specific responses to a foreign antigen or pathogen. A key feature of adaptive immunity is that following the initial contact with antigen (immunologic priming), subsequent antigen exposure leads to more rapid and vigorous immune responses (immunologic memory). The adaptive immune system consists of dual limbs of cellular and humoral immunity. The principal effectors of cellular immunity are T lymphocytes, whereas the principal effectors of humoral immunity are B lymphocytes. Both B and T lymphocytes derive from a common stem cell (Fig. 372e-6).
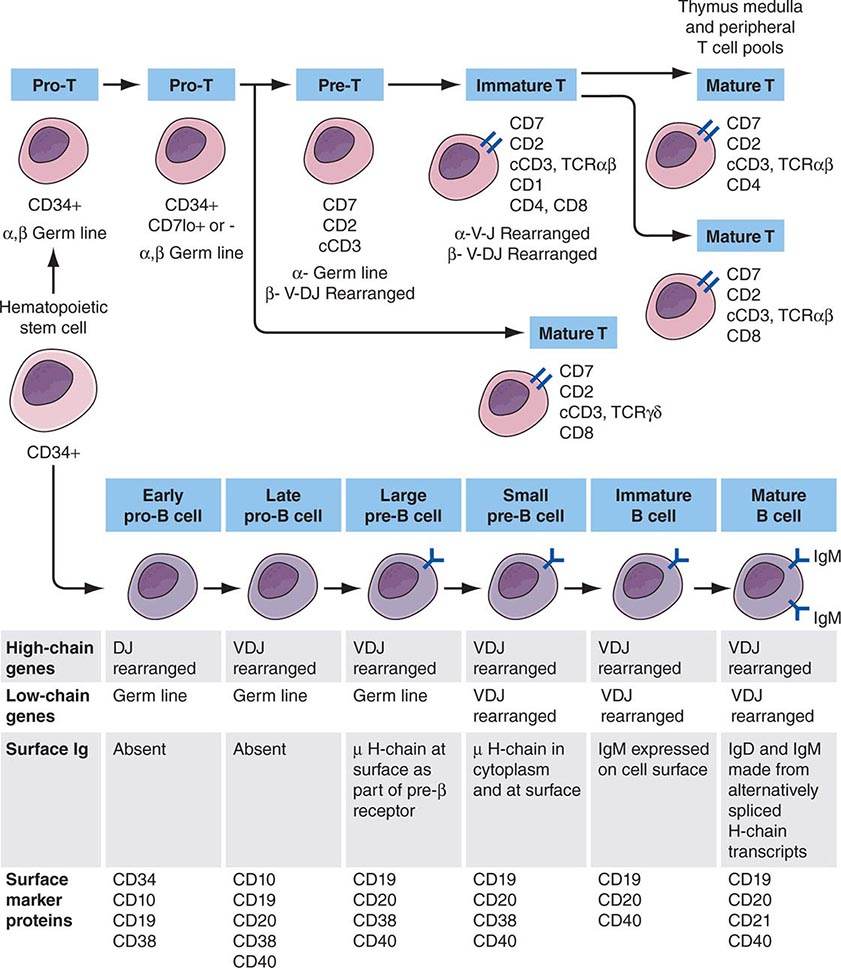
FIGURE 372e-6 Development stages of T and B cells. Elements of the developing T and B cell receptor for antigen are shown schematically. The classification into the various stages of B cell development is primarily defined by rearrangement of the immunoglobulin (Ig) heavy (H) and light (L) chain genes and by the absence or presence of specific surface markers. The classification of stages of T cell development is primarily defined by cell-surface marker protein expression (sCD3, surface CD3 expression; cCD3, cytoplasmic CD3 expression; TCR, T cell receptor). (Adapted from CA Janeway et al [eds]: Immunobiology. The Immune Systemic Health and Disease, 4th ed. New York, Garland, 1999; with permission.)
The proportion and distribution of immunocompetent cells in various tissues reflect cell traffic, homing patterns, and functional capabilities. Bone marrow is the major site of maturation of B cells, monocytes-macrophages, DCs, and granulocytes and contains pluripotent stem cells that, under the influence of various colony-stimulating factors, are capable of giving rise to all hematopoietic cell types. T cell precursors also arise from hematopoietic stem cells and home to the thymus for maturation. Mature T lymphocytes, B lymphocytes, monocytes, and DCs enter the circulation and home to peripheral lymphoid organs (lymph nodes, spleen) and mucosal surface-associated lymphoid tissue (gut, genitourinary, and respiratory tracts) as well as the skin and mucous membranes and await activation by foreign antigen.
T Cells The pool of effector T cells is established in the thymus early in life and is maintained throughout life both by new T cell production in the thymus and by antigen-driven expansion of virgin peripheral T cells into “memory” T cells that reside in peripheral lymphoid organs. The thymus exports ~2% of the total number of thymocytes per day throughout life, with the total number of daily thymic emigrants decreasing by ~3% per year during the first four decades of life.
Mature T lymphocytes constitute 70–80% of normal peripheral blood lymphocytes (only 2% of the total-body lymphocytes are contained in peripheral blood), 90% of thoracic duct lymphocytes, 30–40% of lymph node cells, and 20–30% of spleen lymphoid cells. In lymph nodes, T cells occupy deep paracortical areas around B cell germinal centers, and in the spleen, they are located in periarteriolar areas of white pulp (Chap. 79). T cells are the primary effectors of cell-mediated immunity, with subsets of T cells maturing into CD8+ cytotoxic T cells capable of lysis of virus-infected or foreign cells (short-lived effector T cells) and CD4+ T cells capable of T cell help for CD8+ T cell and B cell development. Two populations of long-lived memory T cells are triggered by infections: effector memory and central memory T cells. Effector memory T cells reside in nonlymphoid organs and respond rapidly to repeated pathogenic infections with cytokine production and cytotoxic functions to kill virus-infected cells. Central memory T cells home to lymphoid organs where they replenish long- and short-lived and effector memory T cells as needed.
In general, CD4+ T cells are the primary regulatory cells of T and B lymphocyte and monocyte function by the production of cytokines and by direct cell contact (Fig. 372e-2). In addition, T cells regulate erythroid cell maturation in bone marrow and, through cell contact (CD40 ligand), have an important role in activation of B cells and induction of Ig isotype switching. Considerable evidence now exists that colonization of the gut by commensal bacteria (the gut microbiome) is responsible for expansion of the peripheral CD4+ T cell compartment in normal children and adults.
Human T cells express cell-surface proteins that mark stages of intrathymic T cell maturation or identify specific functional subpopulations of mature T cells. Many of these molecules mediate or participate in important T cell functions (Table 372e-1, Fig. 372e-6).
The earliest identifiable T cell precursors in bone marrow are CD34+ pro-T cells (i.e., cells in which TCR genes are neither rearranged nor expressed). In the thymus, CD34+ T cell precursors begin cytoplasmic (c) synthesis of components of the CD3 complex of TCR-associated molecules (Fig. 372e-6). Within T cell precursors, TCR for antigen gene rearrangement yields two T cell lineages, expressing either TCR-αβ chains or TCR-γδ chains. T cells expressing the TCR-αβ chains constitute the majority of peripheral T cells in blood, lymph node, and spleen and terminally differentiate into either CD4+ or CD8+ cells. Cells expressing TCR-γδ chains circulate as a minor population in blood; their functions, although not fully understood, have been postulated to be those of immune surveillance at epithelial surfaces and cellular defenses against mycobacterial organisms and other intracellular bacteria through recognition of bacterial lipids.
In the thymus, the recognition of self-peptides on thymic epithelial cells, thymic macrophages, and DCs plays an important role in shaping the T cell repertoire to recognize foreign antigen (positive selection) and in eliminating highly autoreactive T cells (negative selection). As immature cortical thymocytes begin to express surface TCR for antigen, autoreactive thymocytes are destroyed (negative selection), thymocytes with TCRs capable of interacting with foreign antigen peptides in the context of self-MHC antigens are activated and develop to maturity (positive selection), and thymocytes with TCRs that are incapable of binding to self-MHC antigens die of attrition (no selection). Mature thymocytes that are positively selected are either CD4+ helper T cells or MHC class II–restricted cytotoxic (killer) T cells, or they are CD8+ T cells destined to become MHC class I–restricted cytotoxic T cells. MHC class I– or class II–restricted means that T cells recognize antigen peptide fragments only when they are presented in the antigen-recognition site of a class I or class II MHC molecule, respectively (Chap. 373e).
After thymocyte maturation and selection, CD4 and CD8 thymocytes leave the thymus and migrate to the peripheral immune system. The thymus continues to be a contributor to the peripheral immune system well into adult life, both normally and when the peripheral T cell pool is damaged, such as occurs in AIDS and cancer chemotherapy.
MOLECULAR BASIS OF T CELL RECOGNITION OF ANTIGEN The TCR for antigen is a complex of molecules consisting of an antigen-binding heterodimer of either αβ or γδ chains noncovalently linked with five CD3 subunits (γ, δ, ε, ζ, and η) (Fig. 372e-7). The CD3 ζ chains are either disulfide-linked homodimers (CD3-ζ2) or disulfide-linked heterodimers composed of one ζ chain and one η chain. TCR-αβ or TCR-γδ molecules must be associated with CD3 molecules to be inserted into the T cell-surface membrane, TCRα being paired with TCR-β and TCR-γ being paired with TCR-δ. Molecules of the CD3 complex mediate transduction of T cell activation signals via TCRs, whereas TCR-α and -β or -γ and -δ molecules combine to form the TCR antigen-binding site.
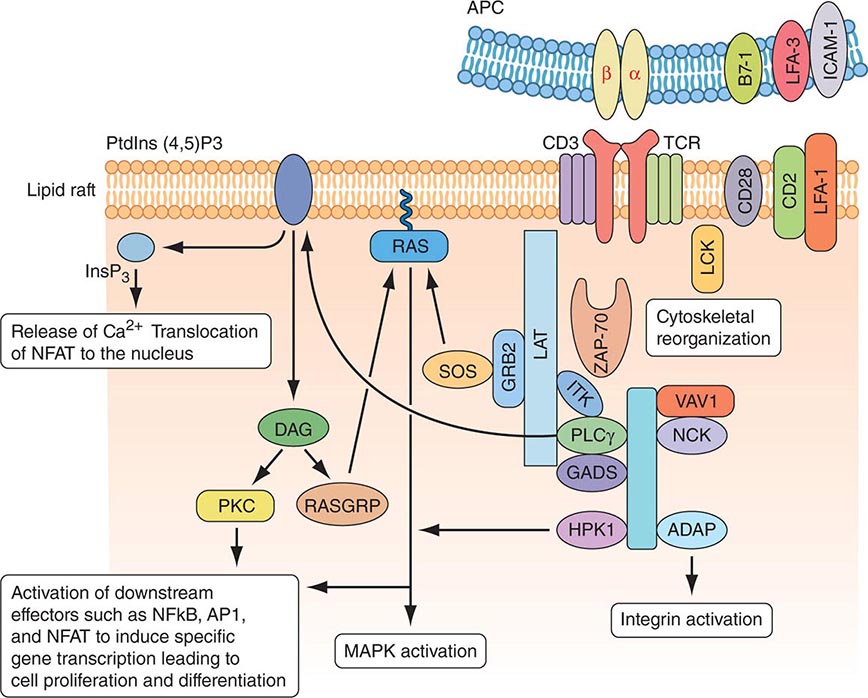
FIGURE 372e-7 Signaling through the T cell receptor. Activation signals are mediated via immunoreceptor tyrosine-based activation (ITAM) sequences in LAT and CD3 chains (blue bars) that bind to enzymes and transduce activation signals to the nucleus via the indicated intracellular activation pathways. Ligation of the T cell receptor (TCR) by MHC complexed with antigen results in sequential activation of LCK and γ-chain-associated protein kinase of 70 kDa (ZAP-70). ZAP-70 phosphorylates several downstream targets, including LAT (linker for activation of T cells) and SLP76 (SCR homology 2 [SH2] domain-containing leukocyte protein of 76 kDa). SLP76 is recruited to membrane-bound LAT through its constitutive interaction with GADS (GRB2-related adaptor protein). Together, SLP76 and LAT nucleate a multimolecular signaling complex, which induces a host of downstream responses, including calcium flux, mitogen-activated protein kinase (MAPK) activation, integrin activation, and cytoskeletal reorganization. APC, antigen-presenting cell. (Adapted from GA Koretzky et al: Nat Rev Immunol 6:67, 2006; with permission from Macmillan Publishers Ltd. Copyright 2006.)
The α, β, γ, and δ TCR for antigen molecules have amino acid sequence homology and structural similarities to immunoglobulin heavy and light chains and are members of the immunoglobulin gene superfamily of molecules. The genes encoding TCR molecules are encoded as clusters of gene segments that rearrange during the course of T cell maturation. This creates an efficient and compact mechanism for housing the diversity requirements of antigen receptor molecules. The TCR-α chain is on chromosome 14 and consists of a series of V (variable), J (joining), and C (constant) regions. The TCR-β chain is on chromosome 7 and consists of multiple V, D (diversity), J, and C TCR-β loci. The TCR-γ chain is on chromosome 7, and the TCR-δ chain is in the middle of the TCR-α locus on chromosome 14. Thus, molecules of the TCR for antigen have constant (framework) and variable regions, and the gene segments encoding the α, β, γ, and δ chains of these molecules are recombined and selected in the thymus, culminating in synthesis of the completed molecule. In both T and B cell precursors (see below), DNA rearrangements of antigen receptor genes involve the same enzymes, recombinase activating gene (RAG) 1 and RAG2, both DNA-dependent protein kinases.
TCR diversity is created by the different V, D, and J segments that are possible for each receptor chain by the many permutations of V, D, and J segment combinations, by “N-region diversification” due to the addition of nucleotides at the junction of rearranged gene segments, and by the pairing of individual chains to form a TCR dimer. As T cells mature in the thymus, the repertoire of antigen-reactive T cells is modified by selection processes that eliminate many autoreactive T cells, enhance the proliferation of cells that function appropriately with self-MHC molecules and antigen, and allow T cells with nonproductive TCR rearrangements to die.
TCR-αβ cells do not recognize native protein or carbohydrate antigens. Instead, T cells recognize only short (~9–13 amino acids) peptide fragments derived from protein antigens taken up or produced in APCs. Foreign antigens may be taken up by endocytosis into acidified intracellular vesicles or by phagocytosis and degraded into small peptides that associate with MHC class II molecules (exogenous antigen-presentation pathway). Other foreign antigens arise endogenously in the cytosol (such as from replicating viruses) and are broken down into small peptides that associate with MHC class I molecules (endogenous antigen-presenting pathway). Thus, APCs proteolytically degrade foreign proteins and display peptide fragments embedded in the MHC class I or II antigen-recognition site on the MHC molecule surface, where foreign peptide fragments are available to bind to TCR-αβ or TCR-γδ chains of reactive T cells. CD4 molecules act as adhesives and, by direct binding to MHC class II (DR, DQ, or DP) molecules, stabilize the interaction of TCR with peptide antigen (Fig. 372e-7). Similarly, CD8 molecules also act as adhesives to stabilize the TCR-antigen interaction by direct CD8 molecule binding to MHC class I (A, B, or C) molecules.
Antigens that arise in the cytosol and are processed via the endogenous antigen-presentation pathway are cleaved into small peptides by a complex of proteases called the proteasome. From the proteasome, antigen peptide fragments are transported from the cytosol into the lumen of the endoplasmic reticulum by a heterodimeric complex termed transporters associated with antigen processing, or TAP proteins. There, MHC class I molecules in the endoplasmic reticulum membrane physically associate with processed cytosolic peptides. Following peptide association with class I molecules, peptide-class I complexes are exported to the Golgi apparatus, and then to the cell surface, for recognition by CD8+ T cells.
Antigens taken up from the extracellular space via endocytosis into intracellular acidified vesicles are degraded by vesicle proteases into peptide fragments. Intracellular vesicles containing MHC class II molecules fuse with peptide-containing vesicles, thus allowing peptide fragments to physically bind to MHC class II molecules. Peptide-MHC class II complexes are then transported to the cell surface for recognition by CD4+ T cells (Chap. 373e).
Whereas it is generally agreed that the TCR-αβ receptor recognizes peptide antigens in the context of MHC class I or class II molecules, lipids in the cell wall of intracellular bacteria such as M. tuberculosis can also be presented to a wide variety of T cells, including subsets of TCR-γδ T cells, and a subset of CD8+ TCR-αβ T cells. Importantly, bacterial lipid antigens are not presented in the context of MHC class I or II molecules, but rather are presented in the context of MHC-related CD1 molecules. Some γδ T cells that recognize lipid antigens via CD1 molecules have very restricted TCR usage, do not need antigen priming to respond to bacterial lipids, and may actually be a form of innate rather than acquired immunity to intracellular bacteria.
Just as foreign antigens are degraded and their peptide fragments presented in the context of MHC class I or class II molecules on APCs, endogenous self-proteins also are degraded, and self-peptide fragments are presented to T cells in the context of MHC class I or class II molecules on APCs. In peripheral lymphoid organs, there are T cells that are capable of recognizing self-protein fragments but normally are anergic or tolerant, i.e., nonresponsive to self-antigenic stimulation, due to lack of self-antigen upregulating APC co-stimulatory molecules such as B7-1 (CD80) and B7-2 (CD86) (see below).
Once engagement of mature T cell TCR by foreign peptide occurs in the context of self-MHC class I or class II molecules, binding of non-antigen-specific adhesion ligand pairs such as CD54-CD11/CD18 and CD58-CD2 stabilizes MHC peptide-TCR binding, and the expression of these adhesion molecules is upregulated (Fig. 372e-7). Once antigen ligation of the TCR occurs, the T cell membrane is partitioned into lipid membrane microdomains, or lipid rafts, that coalesce the key signaling molecules TCR/CD3 complex, CD28, CD2, LAT (linker for activation of T cells), intracellular activated (dephosphorylated) src family protein tyrosine kinases (PTKs), and the key CD3ζ-associated protein-70 (ZAP-70) PTK (Fig. 372e-7). Importantly, during T cell activation, the CD45 molecule, with protein tyrosine phosphatase activity, is partitioned away from the TCR complex to allow activating phosphorylation events to occur. The coalescence of signaling molecules of activated T lymphocytes in microdomains has suggested that T cell-APC interactions can be considered immunologic synapses, analogous in function to neuronal synapses.
After TCR-MHC binding is stabilized, activation signals are transmitted through the cell to the nucleus and lead to the expression of gene products important in mediating the wide diversity of T cell functions such as the secretion of IL-2. The TCR does not have intrinsic signaling activity but is linked to a variety of signaling pathways via immunoreceptor tyrosine-based activation motifs (ITAMs) expressed on the various CD3 chains that bind to proteins that mediate signal transduction. Each of the pathways results in the activation of particular transcription factors that control the expression of cytokine and cytokine receptor genes. Thus, antigen-MHC binding to the TCR induces the activation of the src family of PTKs, fyn and lck (lck is associated with CD4 or CD8 co-stimulatory molecules); phosphorylation of CD3ζ chain; activation of the related tyrosine kinases ZAP-70 and syk; and downstream activation of the calcium-dependent calcineurin pathway, the ras pathway, and the protein kinase C pathway. Each of these pathways leads to activation of specific families of transcription factors (including NF-AT, fos and jun, and rel/NF-κB) that form heteromultimers capable of inducing expression of IL-2, IL-2 receptor, IL-4, TNF-α, and other T cell mediators.
In addition to the signals delivered to the T cell from the TCR complex and CD4 and CD8, molecules on the T cell, such as CD28 and inducible co-stimulator (ICOS), and molecules on DCs, such as B7-1 (CD80) and B7-2 (CD86), also deliver important co-stimulatory signals that upregulate T cell cytokine production and are essential for T cell activation. If signaling through CD28 or ICOS does not occur, or if CD28 is blocked, the T cell becomes anergic rather than activated (see “Immune Tolerance and Autoimmunity” below). CTLA-4 (CD152) is similar to CD28 in its ability to bind CD80 and CD86. Unlike CD28, CTLA-4 transmits an inhibitory signal to T cells, acting as an off switch.
T CELL EXHAUSTION IN VIRAL INFECTIONS AND CANCER In chronic viral infections such as HIV-1, hepatitis C virus, and hepatitis B virus and in chronic malignancies, the persistence of antigen disrupts memory T cell function, resulting in defects in memory T cell responses. This has been defined as T cell exhaustion and is associated with T cell programmed cell death protein 1 (PD-1) (CD279) expression. Exhausted T cells have compromised proliferation and lose the ability to produce effector molecules, like IL-2, TNF-α, and IFN-γ. PD-1 downregulates T cell responses and is associated with T cell exhaustion and disease progression. For this reason, inhibition of T cell PD-1 activity to enhance effector T cell function is being explored as a target for immunotherapy in both viral infections and certain malignancies.
T CELL SUPERANTIGENS Conventional antigens bind to MHC class I or II molecules in the groove of the αβ heterodimer and bind to T cells via the V regions of the TCR-α and -β chains. In contrast, superantigens bind directly to the lateral portion of the TCR-β chain and MHC class II β chain and stimulate T cells based solely on the Vβ gene segment used independent of the D, J, and Vα sequences present. Superantigens are protein molecules capable of activating up to 20% of the peripheral T cell pool, whereas conventional antigens activate <1 in 10,000 T cells. T cell superantigens include staphylococcal enterotoxins and other bacterial products. Superantigen stimulation of human peripheral T cells occurs in the clinical setting of staphylococcal toxic shock syndrome, leading to massive overproduction of T cell cytokines that leads to hypotension and shock (Chap. 172).
B CELLS Mature B cells constitute 10–15% of human peripheral blood lymphocytes, 20–30% of lymph node cells, 50% of splenic lymphocytes, and ~10% of bone marrow lymphocytes. B cells express on their surface intramembrane immunoglobulin (Ig) molecules that function as B cell receptors (BCRs) for antigen in a complex of Ig-associated α and β signaling molecules with properties similar to those described in T cells (Fig. 372e-8). Unlike T cells, which recognize only processed peptide fragments of conventional antigens embedded in the notches of MHC class I and class II antigens of APCs, B cells are capable of recognizing and proliferating to whole unprocessed native antigens via antigen binding to B cell–surface Ig (sIg) receptors. B cells also express surface receptors for the Fc region of IgG molecules (CD32) as well as receptors for activated complement components (C3d or CD21, C3b or CD35). The primary function of B cells is to produce antibodies. B cells also serve as APCs and are highly efficient at antigen processing. Their antigen-presenting function is enhanced by a variety of cytokines. Mature B cells are derived from bone marrow precursor cells that arise continuously throughout life (Fig. 372e-6).
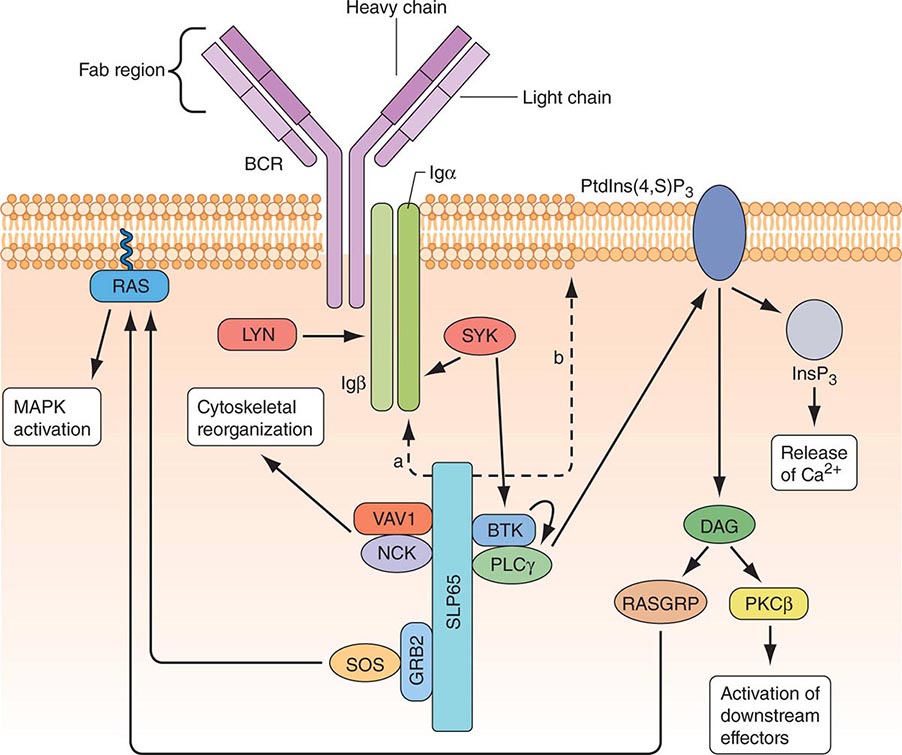
FIGURE 372e-8 B cell receptor (BCR) activation results in the sequential activation of protein tyrosine kinases, which results in the formation of a signaling complex and activation of downstream pathways as shown. Whereas SLP76 is recruited to the membrane through GADS and LAT, the mechanism of SLP65 recruitment is unclear. Studies have indicated two mechanisms: (a) direct binding by the SH2 domain of SLP65 to immunoglobulin (Ig) of the BCR complex or (b) membrane recruitment through a leucine zipper in the amino terminus of SLP65 and an unknown binding partner. ADAP, adhesion- and degranulation-promoting adaptor protein; AP1, activator protein 1; BTK, Bruton’s tyrosine kinase; DAG, diacylglycerol; GRB2, growth factor receptor-bound protein 2; HPK1, hematopoietic progenitor kinase 1; InsP3, inositol-1,4,5-trisphosphate; ITK, interleukin-2-inducible T cell kinase; NCK, noncatalytic region of tyrosine kinase; NF-B, nuclear factor B; PKC, protein kinase C; PLC, phospholipase C; PtdIns(4,5)P2, phosphatidylinositol-4,5-bisphosphate; RASGRP, RAS guanyl-releasing protein; SOS, son of sevenless homologue; SYK, spleen tyrosine kinase. (Adapted from GA Koretzky et al: Nat Rev Immunol 6:67, 2006; with permission from Macmillan Publishers Ltd. Copyright 2006.)
B lymphocyte development can be separated into antigen-independent and antigen-dependent phases. Antigen-independent B cell development occurs in primary lymphoid organs and includes all stages of B cell maturation up to the sIg+ mature B cell. Antigen-dependent B cell maturation is driven by the interaction of antigen with the mature B cell sIg, leading to memory B cell induction, Ig class switching, and plasma cell formation. Antigen-dependent stages of B cell maturation occur in secondary lymphoid organs, including lymph node, spleen, and gut Peyer’s patches. In contrast to the T cell repertoire that is generated intrathymically before contact with foreign antigen, the repertoire of B cells expressing diverse antigen-reactive sites is modified by further alteration of Ig genes after stimulation by antigen—a process called somatic hypermutation—that occurs in lymph node germinal centers.
During B cell development, diversity of the antigen-binding variable region of Ig is generated by an ordered set of Ig gene rearrangements that are similar to the rearrangements undergone by TCR α, β, γ, and δ genes. For the heavy chain, there is first a rearrangement of D segments to J segments, followed by a second rearrangement between a V gene segment and the newly formed D-J sequence; the C segment is aligned to the V-D-J complex to yield a functional Ig heavy chain gene (V-D-J-C). During later stages, a functional κ or γ light chain gene is generated by rearrangement of a V segment to a J segment, ultimately yielding an intact Ig molecule composed of heavy and light chains.
The process of Ig gene rearrangement is regulated and results in a single antibody specificity produced by each B cell, with each Ig molecule comprising one type of heavy chain and one type of light chain. Although each B cell contains two copies of Ig light and heavy chain genes, only one gene of each type is productively rearranged and expressed in each B cell, a process termed allelic exclusion.
There are ~300 Vκ genes and 5 Jκ genes, resulting in the pairing of Vκ and Jκ genes to create >1500 different kappa light chain combinations. There are ~70 Vλ genes and 4Jλ genes for >280 different lambda light chain combinations. The number of distinct light chains that can be generated is increased by somatic mutations within the V and J genes, thus creating large numbers of possible specificities from a limited amount of germline genetic information. As noted above, in heavy chain Ig gene rearrangement, the VH domain is created by the joining of three types of germline genes called VH, DH, and JH, thus allowing for even greater diversity in the variable region of heavy chains than of light chains.
The most immature B cell precursors (early pro-B cells) lack cytoplasmic Ig (cIg) and sIg (Fig. 372e-6). The large pre-B cell is marked by the acquisition of the surface pre-BCR composed of μ heavy (H) chains and a pre-B light chain, termed ψLC. ψLC is a surrogate light chain receptor encoded by the nonrearranged V pre-B and the γ5 light chain locus (the pre-BCR). Pro- and pre-B cells are driven to proliferate and mature by signals from bone marrow stroma—in particular, IL-7. Light chain rearrangement occurs in the small pre-B cell stage such that the full BCR is expressed at the immature B cell stage. Immature B cells have rearranged Ig light chain genes and express sIgM. As immature B cells develop into mature B cells, sIgD is expressed as well as sIgM. At this point, B lineage development in bone marrow is complete, and B cells exit into the peripheral circulation and migrate to secondary lymphoid organs to encounter specific antigens.
Random rearrangements of Ig genes occasionally generate self-reactive antibodies, and mechanisms must be in place to correct these mistakes. One such mechanism is BCR editing, whereby autoreactive BCRs are mutated to not react with self-antigens. If receptor editing is unsuccessful in eliminating autoreactive B cells, then autoreactive B cells undergo negative selection in the bone marrow through induction of apoptosis after BCR engagement of self-antigen.
After leaving the bone marrow, B cells populate peripheral B cell sites, such as lymph node and spleen, and await contact with foreign antigens that react with each B cell’s clonotypic receptor. Antigen-driven B cell activation occurs through the BCR, and a process known as somatic hypermutation takes place whereby point mutations in rearranged H- and L-genes give rise to mutant sIg molecules, some of which bind antigen better than the original sIg molecules. Somatic hypermutation, therefore, is a process whereby memory B cells in peripheral lymph organs have the best binding, or the highest-affinity antibodies. This overall process of generating the best antibodies is called affinity maturation of antibody.
Lymphocytes that synthesize IgG, IgA, and IgE are derived from sIgM+, sIgD+ mature B cells. Ig class switching occurs in lymph node and other peripheral lymphoid tissue germinal centers. CD40 on B cells and CD40 ligand on T cells constitute a critical co-stimulatory receptor-ligand pair of immune-stimulatory molecules. Pairs of CD40+ B cells and CD40 ligand+ T cells bind and drive B cell Ig class switching via T cell-produced cytokines such as IL-4 and TGF-β. IL-1, -2, -4, -5, and -6 synergize to drive mature B cells to proliferate and differentiate into Ig-secreting cells.
Humoral Mediators of Adaptive Immunity: Immunoglobulins Immunoglobulins are the products of differentiated B cells and mediate the humoral arm of the immune response. The primary functions of antibodies are to bind specifically to antigen and bring about the inactivation or removal of the offending toxin, microbe, parasite, or other foreign substance from the body. The structural basis of Ig molecule function and Ig gene organization has provided insight into the role of antibodies in normal protective immunity, pathologic immune-mediated damage by immune complexes, and autoantibody formation against host determinants.
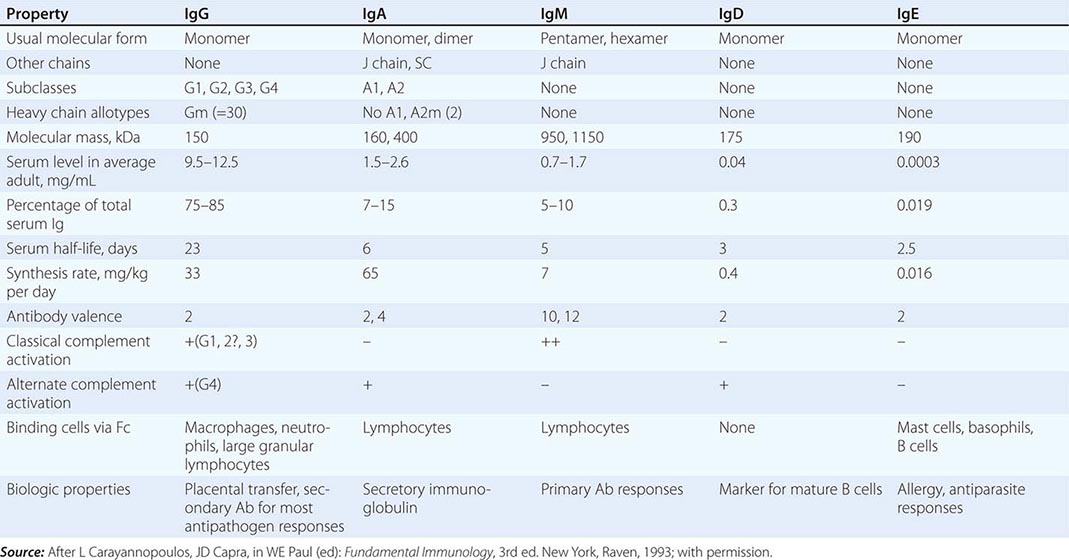
All immunoglobulins have the basic structure of two heavy and two light chains (Fig. 372e-8). Immunoglobulin isotype (i.e., G, M, A, D, E) is determined by the type of Ig heavy chain present. IgG and IgA isotypes can be divided further into subclasses (G1, G2, G3, G4, and A1, A2) based on specific antigenic determinants on Ig heavy chains. The characteristics of human immunoglobulins are outlined in Table 372e-12. The four chains are covalently linked by disulfide bonds. Each chain is made up of a V region and C regions (also called domains), themselves made up of units of ~110 amino acids. Light chains have one variable (VL) and one constant (CL) unit; heavy chains have one variable unit (VH) and three or four constant (CH) units, depending on isotype. As the name suggests, the constant, or C, regions of Ig molecules are made up of homologous sequences and share the same primary structure as all other Ig chains of the same isotype and subclass. Constant regions are involved in biologic functions of Ig molecules. The CH2 domain of IgG and the CH4 units of IgM are involved with the binding of the C1q portion of C1 during complement activation. The CH region at the carboxy-terminal end of the IgG molecule, the Fc region, binds to surface Fc receptors (CD16, CD32, CD64) of macrophages, DCs, NK cells, B cells, neutrophils, and eosinophils. The Fc of IgA binds to FcαR (CD89), and the Fc of IgE binds to FcεR (CD23).
PHYSICAL, CHEMICAL, AND BIOLOGIC PROPERTIES OF HUMAN IMMUNOGLOBULINS |
Variable regions (VL and VH) constitute the antibody-binding (Fab) region of the molecule. Within the VL and VH regions are hypervariable regions (extreme sequence variability) that constitute the antigen-binding site unique to each Ig molecule. The idiotype is defined as the specific region of the Fab portion of the Ig molecule to which antigen binds. Antibodies against the idiotype portion of an antibody molecule are called anti-idiotype antibodies. The formation of such antibodies in vivo during a normal B cell antibody response may generate a negative (or “off”) signal to B cells to terminate antibody production.
IgG constitutes ~75–85% of total serum immunoglobulin. The four IgG subclasses are numbered in order of their level in serum, IgG1 being found in greatest amounts and IgG4 the least. IgG subclasses have clinical relevance in their varying ability to bind macrophage and neutrophil Fc receptors and to activate complement (Table 372e-12). Moreover, selective deficiencies of certain IgG subclasses give rise to clinical syndromes in which the patient is inordinately susceptible to bacterial infections. IgG antibodies are frequently the predominant antibody made after rechallenge of the host with antigen (secondary antibody response).
IgM antibodies normally circulate as a 950-kDa pentamer with 160-kDa bivalent monomers joined by a molecule called the J chain, a 15-kDa nonimmunoglobulin molecule that also effects polymerization of IgA molecules. IgM is the first immunoglobulin to appear in the immune response (primary antibody response) and is the initial type of antibody made by neonates. Membrane IgM in the monomeric form also functions as a major antigen receptor on the surface of mature B cells (Table 372e-12). IgM is an important component of immune complexes in autoimmune diseases. For example, IgM antibodies against IgG molecules (rheumatoid factors) are present in high titers in rheumatoid arthritis, other collagen diseases, and some infectious diseases (subacute bacterial endocarditis).
IgA constitutes only 7–15% of total serum immunoglobulin but is the predominant class of immunoglobulin in secretions. IgA in secretions (tears, saliva, nasal secretions, gastrointestinal tract fluid, and human milk) is in the form of secretory IgA (sIgA), a polymer consisting of two IgA monomers, a joining molecule, again called the J chain, and a glycoprotein called the secretory protein. Of the two IgA subclasses, IgA1 is primarily found in serum, whereas IgA2 is more prevalent in secretions. IgA fixes complement via the alternative complement pathway and has potent antiviral activity in humans by prevention of virus binding to respiratory and gastrointestinal epithelial cells.
IgD is found in minute quantities in serum and, together with IgM, is a major receptor for antigen on the naïve B cell surface. IgE, which is present in serum in very low concentrations, is the major class of immunoglobulin involved in arming mast cells and basophils by binding to these cells via the Fc region. Antigen cross-linking of IgE molecules on basophil and mast cell surfaces results in release of mediators of the immediate hypersensitivity (allergic) response (Table 372e-12).
CELLULAR INTERACTIONS IN REGULATION OF NORMAL IMMUNE RESPONSES
The net result of activation of the humoral (B cell) and cellular (T cell) arms of the adaptive immune system by foreign antigen is the elimination of antigen directly by specific effector T cells or in concert with specific antibody. Figure 372e-2 is a simplified schematic diagram of the T and B cell responses indicating some of these cellular interactions.
The expression of adaptive immune cell function is the result of a complex series of immunoregulatory events that occur in phases. Both T and B lymphocytes mediate immune functions, and each of these cell types, when given appropriate signals, passes through stages, from activation and induction through proliferation, differentiation, and ultimately effector functions. The effector function expressed may be at the end point of a response, such as secretion of antibody by a differentiated plasma cell, or it might serve a regulatory function that modulates other functions, such as is seen with CD4+ and CD8+ T lymphocytes that modulate both differentiation of B cells and activation of CD8+ cytotoxic T cells.
CD4 helper T cells can be subdivided on the basis of cytokines produced (Fig. 372e-2). Activated TH1-type helper T cells secrete IL-2, IFN-γ, IL-3, TNF-α, GM-CSF, and TNF-β, whereas activated TH2-type helper T cells secrete IL-3, -4, -5, -6, -10, and -13. TH1 CD4+ T cells, through elaboration of IFN-γ, have a central role in mediating intracellular killing by a variety of pathogens. TH1 CD4+ T cells also provide T cell help for generation of cytotoxic T cells and some types of opsonizing antibody, and they generally respond to antigens that lead to delayed hypersensitivity types of immune responses for many intracellular viruses and bacteria (such as HIV or M. tuberculosis). In contrast, TH2 cells have a primary role in regulatory humoral immunity and isotype switching. TH2 cells, through production of IL-4 and IL-10, have a regulatory role in limiting proinflammatory responses mediated by TH1 cells (Fig. 372e-2). In addition, TH2 CD4+ T cells provide help to B cells for specific Ig production and respond to antigens that require high antibody levels for foreign antigen elimination (extracellular encapsulated bacteria such as Streptococcus pneumoniae and certain parasite infections). A new subset of the TH family has been described, termed TH17, characterized as cells that secrete cytokines such as IL-17, -22, and -26. TH17 cells have been shown to play a role in autoimmune inflammatory disorders in addition to defense against extracellular bacteria and fungi, particularly at mucosal surfaces. In summary, the type of T cell response generated in an immune response is determined by the microbe PAMPs presented to the DCs, the TLRs on the DCs that become activated, the types of DCs that are activated, and the cytokines that are produced (Table 372e-4). Commonly, myeloid DCs produce IL-12 and activate TH1 T cell responses that result in IFN-γ and cytotoxic T cell induction, and plasmacytoid DCs produce IFN-α and lead to TH2 responses that result in IL-4 production and enhanced antibody responses.
As shown in Figs. 372e-2 and 372e-3, upon activation by DCs, T cell subsets that produce IL-2, IL-3, IFN-γ, and/or IL-4, -5, -6, -10, and -13 are generated and exert positive and negative influences on effector T and B cells. For B cells, trophic effects are mediated by a variety of cytokines, particularly T cell–derived IL-3, -4, -5, and -6, that act at sequential stages of B cell maturation, resulting in B cell proliferation, differentiation, and ultimately antibody secretion. For cytotoxic T cells, trophic factors include inducer T cell secretion of IL-2, IFN-γ, and IL-12.
An important type of immunomodulatory T cell that controls immune responses is CD4+ and CD8+ T regulatory cells. These cells constitutively express the α chain of the IL-2 receptor (CD25), produce large amounts of IL-10, and can suppress both T and B cell responses. T regulatory cells are induced by immature DCs and play key roles in maintaining tolerance to self-antigens in the periphery. Loss of T regulatory cells is the cause of organ-specific autoimmune disease in mice such as autoimmune thyroiditis, adrenalitis, and oophoritis (see “Immune Tolerance and Autoimmunity” below). T regulatory cells also play key roles in controlling the magnitude and duration of immune responses to microbes. Normally, after the initial immune response to a microbe has eliminated the invader, T regulatory cells are activated to suppress the antimicrobe response and prevent host injury. Some microbes have adapted to induce T regulatory cell activation at the site of infection to promote parasite infection and survival. In Leishmania infection, the parasite locally induces T regulatory cell accumulation at skin infection sites that dampens anti-Leishmania T cell responses and prevents elimination of the parasite. It is thought that many chronic infections such as by M. tuberculosis are associated with abnormal T regulatory cell activation that prevents elimination of the microbe.
Although B cells recognize native antigen via B cell–surface Ig receptors, B cells require T cell help to produce high-affinity antibody of multiple isotypes that are the most effective in eliminating foreign antigen. This T cell dependence likely functions in the regulation of B cell responses and in protection against excessive autoantibody production. T cell–B cell interactions that lead to high-affinity antibody production require (1) processing of native antigen by B cells and expression of peptide fragments on the B cell surface for presentation to TH cells, (2) the ligation of B cells by both the TCR complex and the CD40 ligand, (3) induction of the process termed antibody isotype switching in antigen-specific B cell clones, and (4) induction of the process of affinity maturation of antibody in the germinal centers of B cell follicles of lymph node and spleen.
Naïve B cells express cell-surface IgD and IgM, and initial contact of naïve B cells with antigen is via binding of native antigen to B cell–surface IgM. T cell cytokines, released following TH2 cell contact with B cells or by a “bystander” effect, induce changes in Ig gene conformation that promote recombination of Ig genes. These events then result in the “switching” of expression of heavy chain exons in a triggered B cell, leading to the secretion of IgG, IgA, or, in some cases, IgE antibody with the same V region antigen specificity as the original IgM antibody, for response to a wide variety of extracellular bacteria, protozoa, and helminths. CD40 ligand expression by activated T cells is critical for induction of B cell antibody isotype switching and for B cell responsiveness to cytokines. Patients with mutations in T cell CD40 ligand have B cells that are unable to undergo isotype switching, resulting in lack of memory B cell generation and the immunodeficiency syndrome of X-linked hyper-IgM syndrome (Chap. 374).
IMMUNE TOLERANCE AND AUTOIMMUNITY
Immune tolerance is defined as the absence of activation of pathogenic autoreactivity. Autoimmune diseases are syndromes caused by the activation of T or B cells or both, with no evidence of other causes such as infections or malignancies (Chap. 377e). Once thought to be mutually exclusive, immune tolerance and autoimmunity are now both recognized to be present normally in health; when abnormal, they represent extremes from the normal state. For example, it is now known that low levels of autoreactivity of T and B cells with self-antigens in the periphery are critical to their survival. Similarly, low levels of autoreactivity and thymocyte recognition of self-antigens in the thymus are the mechanisms whereby (1) normal T cells are positively selected to survive and leave the thymus to respond to foreign microbes in the periphery and (2) T cells highly reactive to self-antigens are negatively selected and die to prevent overly self-reactive T cells from getting into the periphery (central tolerance). However, not all self-antigens are expressed in the thymus to delete highly self-reactive T cells, and there are mechanisms for peripheral tolerance induction of T cells as well. Unlike the presentation of microbial antigens by mature DCs, the presentation of self-antigens by immature DCs neither activates nor matures the DCs to express high levels of co-stimulatory molecules such as B7-1 (CD80) or B7-2 (CD86). When peripheral T cells are stimulated by DCs expressing self-antigens in the context of HLA molecules, sufficient stimulation of T cells occurs to keep them alive, but otherwise they remain anergic, or nonresponsive, until they contact a DC with high levels of co-stimulatory molecules expressing microbial antigens. In the latter setting, normal T cells then become activated to respond to the microbe. If B cells have high-self-reactivity BCRs, they normally undergo either deletion in the bone marrow or receptor editing to express a less autoreactive receptor. Although many autoimmune diseases are characterized by abnormal or pathogenic autoantibody production (Table 372e-13), most autoimmune diseases are caused by a combination of excess T and B cell reactivity.
RECOMBINANT OR PURIFIED AUTOANTIGENS RECOGNIZED BY AUTOANTIBODIES ASSOCIATED WITH HUMAN AUTOIMMUNE DISORDERS |
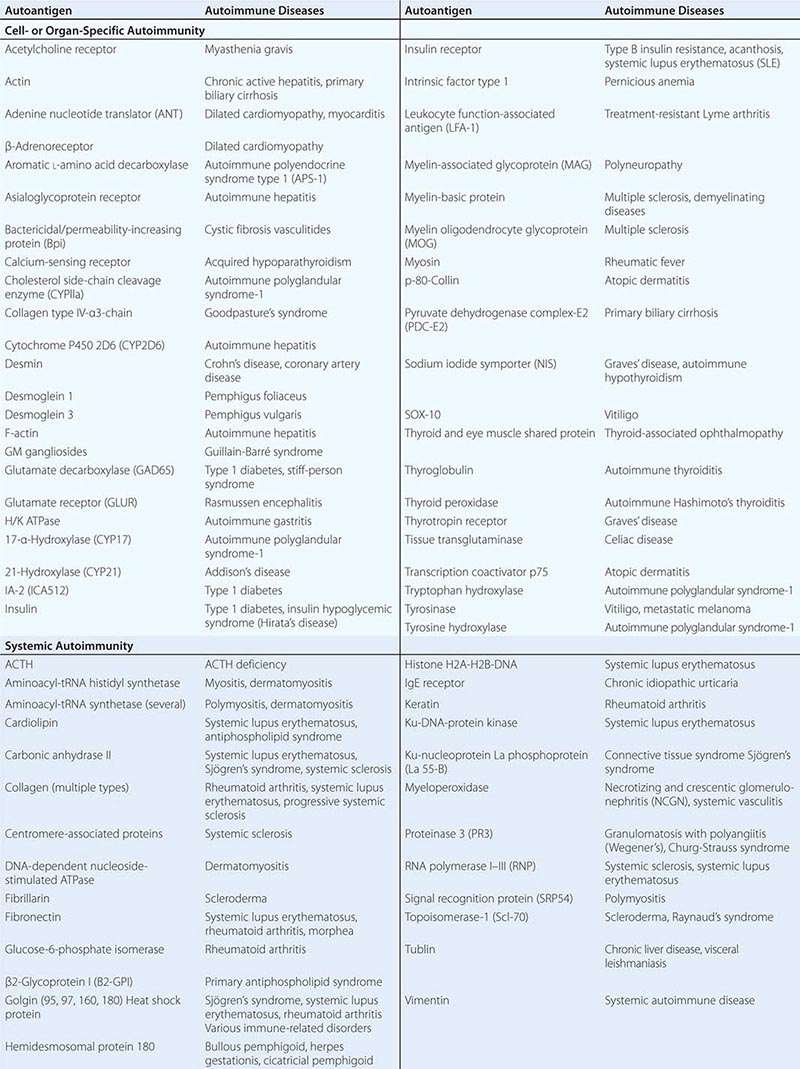
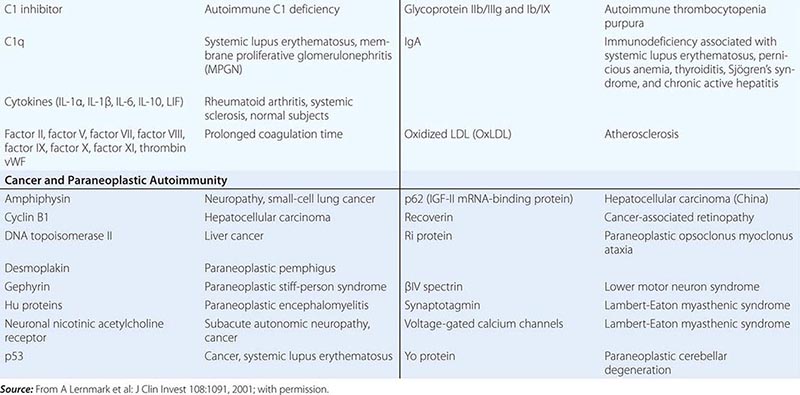
Multiple factors contribute to the genesis of clinical autoimmune disease syndromes, including genetic susceptibility (Table 372e-13), environmental immune stimulants such as drugs (e.g., procainamide and phenytoin [Dilantin] with drug-induced systemic lupus erythematosus), infectious agent triggers (such as Epstein-Barr virus and autoantibody production against red blood cells and platelets), and loss of T regulatory cells (leading to thyroiditis, adrenalitis, and oophoritis).
Immunity at Mucosal Surfaces Mucosa covering the respiratory, digestive, and urogenital tracts; the eye conjunctiva; the inner ear; and the ducts of all exocrine glands contain cells of the innate and adaptive mucosal immune system that protect these surfaces against pathogens. In the healthy adult, mucosa-associated lymphoid tissue (MALT) contains 80% of all immune cells within the body and constitutes the largest mammalian lymphoid organ system.
MALT has three main functions: (1) to protect the mucous membranes from invasive pathogens; (2) to prevent uptake of foreign antigens from food, commensal organisms, and airborne pathogens and particulate matter; and (3) to prevent pathologic immune responses from foreign antigens if they do cross the mucosal barriers of the body (Fig. 372e-9).
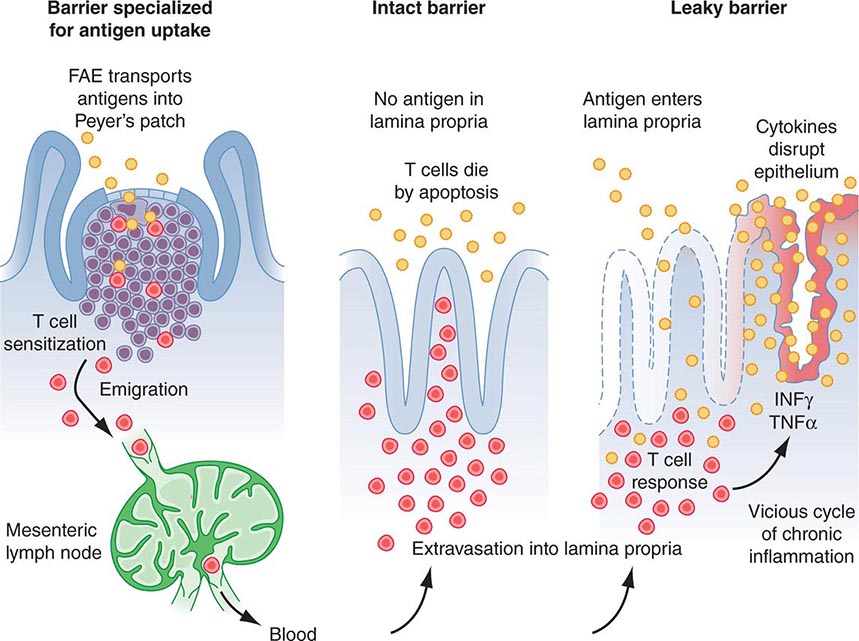
FIGURE 372e-9 Increased epithelial permeability may be important in the development of chronic gut T cell–mediated inflammation. CD4 T cells activated by gut antigens in Peyer’s patches migrate to the lamina propria (LP). In healthy individuals, these cells die by apoptosis. Increased epithelial permeability may allow sufficient antigen to enter the LP to trigger T cell activation, breaking tolerance mediated by immunosuppressive cytokines and perhaps T regulatory cells. Proinflammatory cytokines then further increase epithelial permeability, setting up a vicious cycle of chronic inflammation. (From TT MacDonald et al: Science 307:1920, 2005; with permission.)
MALT is a compartmentalized system of immune cells that functions independently from systemic immune organs. Whereas the systemic immune organs are essentially sterile under normal conditions and respond vigorously to pathogens, MALT immune cells are continuously bathed in foreign proteins and commensal bacteria, and they must select those pathogenic antigens that must be eliminated. MALT contains anatomically defined foci of immune cells in the intestine, tonsil, appendix, and peribronchial areas that are inductive sites for mucosal immune responses. From these sites, immune T and B cells migrate to effector sites in mucosal parenchyma and exocrine glands where mucosal immune cells eliminate pathogen-infected cells. In addition to mucosal immune responses, all mucosal sites have strong mechanical and chemical barriers and cleansing functions to repel pathogens.
Key components of MALT include specialized epithelial cells called “membrane” or “M” cells that take up antigens and deliver them to DCs or other APCs. Effector cells in MALT include B cells producing antipathogen neutralizing antibodies of secretory IgA as well as IgG isotype, T cells producing similar cytokines as in systemic immune system response, and T helper and cytotoxic T cells that respond to pathogen-infected cells.
Secretory IgA is produced in amounts of >50 mg/kg of body weight per 24 h and functions to inhibit bacterial adhesion, inhibit macromolecule absorption in the gut, neutralize viruses, and enhance antigen elimination in tissue through binding to IgA and receptor-mediated transport of immune complexes through epithelial cells.
Recent studies have demonstrated the importance of commensal gut and other mucosal bacteria to the health of the human immune system. Normal commensal flora induces anti-inflammatory events in the gut and protects epithelial cells from pathogens through TLRs and other PRR signaling. When the gut is depleted of normal commensal flora, the immune system becomes abnormal, with loss of TH1 T cell function. Restoration of the normal gut flora can reestablish the balance in T helper cell ratios characteristic of the normal immune system. When the gut barrier is intact, either antigens do not transverse the gut epithelium or, when pathogens are present, a self-limited, protective MALT immune response eliminates the pathogen (Fig. 372e-9). However, when the gut barrier breaks down, immune responses to commensal flora antigens can cause inflammatory bowel diseases such as Crohn’s disease and, perhaps, ulcerative colitis (Fig. 372e-9) (Chap. 351). Uncontrolled MALT immune responses to food antigens, such as gluten, can cause celiac disease (Chap. 351).
THE CELLULAR AND MOLECULAR CONTROL OF PROGRAMMED CELL DEATH
The process of apoptosis (programmed cell death) plays a crucial role in regulating normal immune responses to antigen. In general, a wide variety of stimuli trigger one of several apoptotic pathways to eliminate microbe-infected cells, eliminate cells with damaged DNA, or eliminate activated immune cells that are no longer needed (Fig. 372e-10). The largest known family of “death receptors” is the TNF receptor (TNF-R) family (TNF-R1, TNF-R2, Fas [CD95], death receptor 3 [DR3], death receptor 4 [DR4; TNF-related apoptosis-including ligand receptor 1, or TRAIL-R1], and death receptor 5 [DR5, TRAIL-R2]); their ligands are all in the TNF-α family. Binding of ligands to these death receptors leads to a signaling cascade that involves activation of the caspase family of molecules that leads to DNA cleavage and cell death. Two other pathways of programmed cell death involve nuclear p53 in the elimination of cells with abnormal DNA and mitochondrial cytochrome c to induce cell death in damaged cells (Fig. 372e-10). A number of human diseases have now been described that result from, or are associated with, mutated apoptosis genes (Table 372e-14). These include mutations in the Fas and Fas ligand genes in autoimmune and lymphoproliferation syndromes, and multiple associations of mutations in genes in the apoptotic pathway with malignant syndromes.
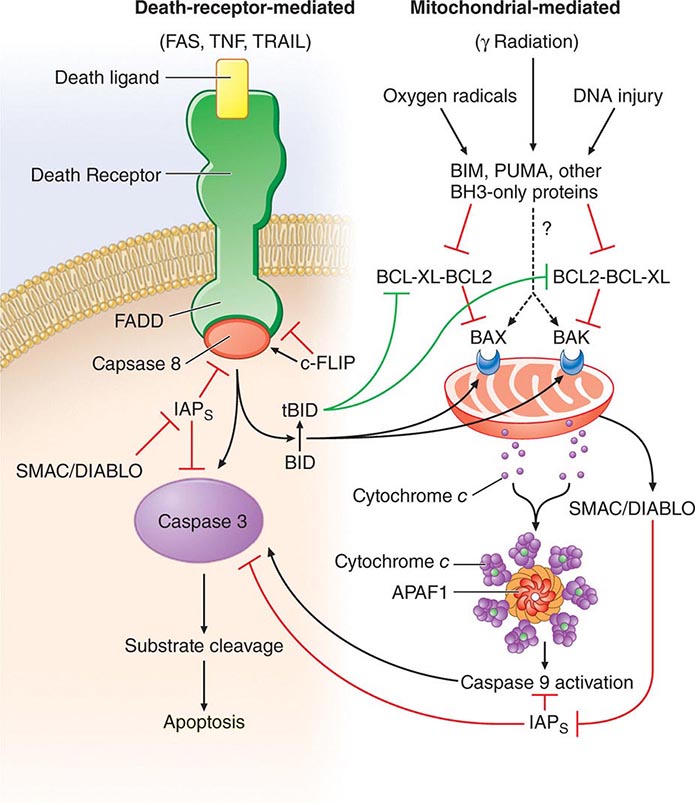
FIGURE 372e-10 Pathways of cellular apoptosis. There are two major pathways of apoptosis: the death-receptor pathway, which is mediated by activation of death receptors, and the BCL2-regulated mitochondrial pathway, which is mediated by noxious stimuli that ultimately lead to mitochondrial injury. Ligation of death receptors recruits the adaptor protein FAS-associated death domain (FADD). FADD in turn recruits caspase 8, which ultimately activates caspase 3, the key “executioner” caspase. Cellular FLICE-inhibitory protein (c-FLIP) can either inhibit or potentiate binding of FADD and caspase 8, depending on its concentration. In the intrinsic pathway, proapoptotic BH3 proteins are activated by noxious stimuli, which interact with and inhibit antiapoptotic BCL2 or BCL-XL. Thus, BAX and BAK are free to induce mitochondrial permeabilization with release of cytochrome c, which ultimately results in the activation of caspase 9 through the apoptosome. Caspase 9 then activates caspase 3. SMAC/DIABLO is also released after mitochondrial permeabilization and acts to block the action of inhibitors of apoptosis protein (IAPs), which inhibit caspase activation. There is potential cross-talk between the two pathways, which is mediated by the truncated form of BID (tBID) that is produced by caspase 8–mediated BID cleavage; tBID acts to inhibit the BCL2-BCL-XL pathway and to activate BAX and BAK. There is debate (indicated by the question mark) as to whether proapoptotic BH3 molecules (e.g., BIM and PUMA) act directly on BAX and BAK to induce mitochondrial permeability or whether they act only on BCL2-BCL-XL. APAF1, apoptotic protease-activating factor 1; BH3, BCL homologue; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand. (From RS Hotchkiss et al: N Engl J Med 361:1570, 2009; with permission.)
IMMUNE SYSTEM MOLECULE DEFECTS IN ANIMALS OR HUMANS THAT CAUSE AUTOIMMUNE OR MALIGNANT SYNDROMES |
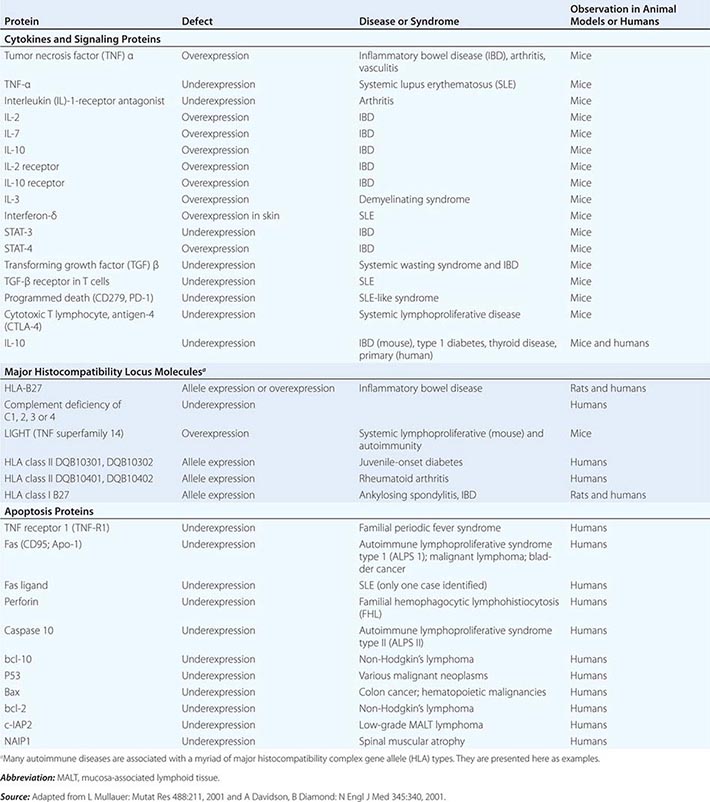
MECHANISMS OF IMMUNE-MEDIATED DAMAGE TO MICROBES OR HOST TISSUES
Several responses by the host innate and adaptive immune systems to foreign microbes culminate in rapid and efficient elimination of microbes. In these scenarios, the classic weapons of the adaptive immune system (T cells, B cells) interface with cells (macrophages, DCs, NK cells, neutrophils, eosinophils, basophils) and soluble products (microbial peptides, pentraxins, complement and coagulation systems) of the innate immune system (Chaps. 80 and 376).
There are five general phases of host defenses: (1) migration of leukocytes to sites of antigen localization; (2) antigen-nonspecific recognition of pathogens by macrophages and other cells and systems of the innate immune system; (3) specific recognition of foreign antigens mediated by T and B lymphocytes; (4) amplification of the inflammatory response with recruitment of specific and nonspecific effector cells by complement components, cytokines, kinins, arachidonic acid metabolites, and mast cell–basophil products; and (5) macrophage, neutrophil, and lymphocyte participation in destruction of antigen with ultimate removal of antigen particles by phagocytosis (by macrophages or neutrophils) or by direct cytotoxic mechanisms (involving macrophages, neutrophils, DCs, and lymphocytes). Under normal circumstances, orderly progression of host defenses through these phases results in a well-controlled immune and inflammatory response that protects the host from the offending antigen. However, dysfunction of any of the host defense systems can damage host tissue and produce clinical disease. Furthermore, for certain pathogens or antigens, the normal immune response itself might contribute substantially to the tissue damage. For example, the immune and inflammatory response in the brain to certain pathogens such as M. tuberculosis may be responsible for much of the morbidity rate of this disease in that organ system (Chap. 202). In addition, the morbidity rate associated with certain pneumonias such as that caused by Pneumocystis jiroveci may be associated more with inflammatory infiltrates than with the tissue-destructive effects of the microorganism itself (Chap. 244).
Molecular Basis of Lymphocyte–Endothelial Cell Interactions The control of lymphocyte circulatory patterns between the bloodstream and peripheral lymphoid organs operates at the level of lymphocyte–endothelial cell interactions to control the specificity of lymphocyte subset entry into organs. Similarly, lymphocyte–endothelial cell interactions regulate the entry of lymphocytes into inflamed tissue. Adhesion molecule expression on lymphocytes and endothelial cells regulates the retention and subsequent egress of lymphocytes within tissue sites of antigenic stimulation, delaying cell exit from tissue and preventing reentry into the circulating lymphocyte pool (Fig. 372e-11). All types of lymphocyte migration begin with lymphocyte attachment to specialized regions of vessels, termed high endothelial venules (HEVs). An important concept is that adhesion molecules do not generally bind their ligand until a conformational change (ligand activation) occurs in the adhesion molecule that allows ligand binding. Induction of a conformation-dependent determinant on an adhesion molecule can be accomplished by cytokines or via ligation of other adhesion molecules on the cell.
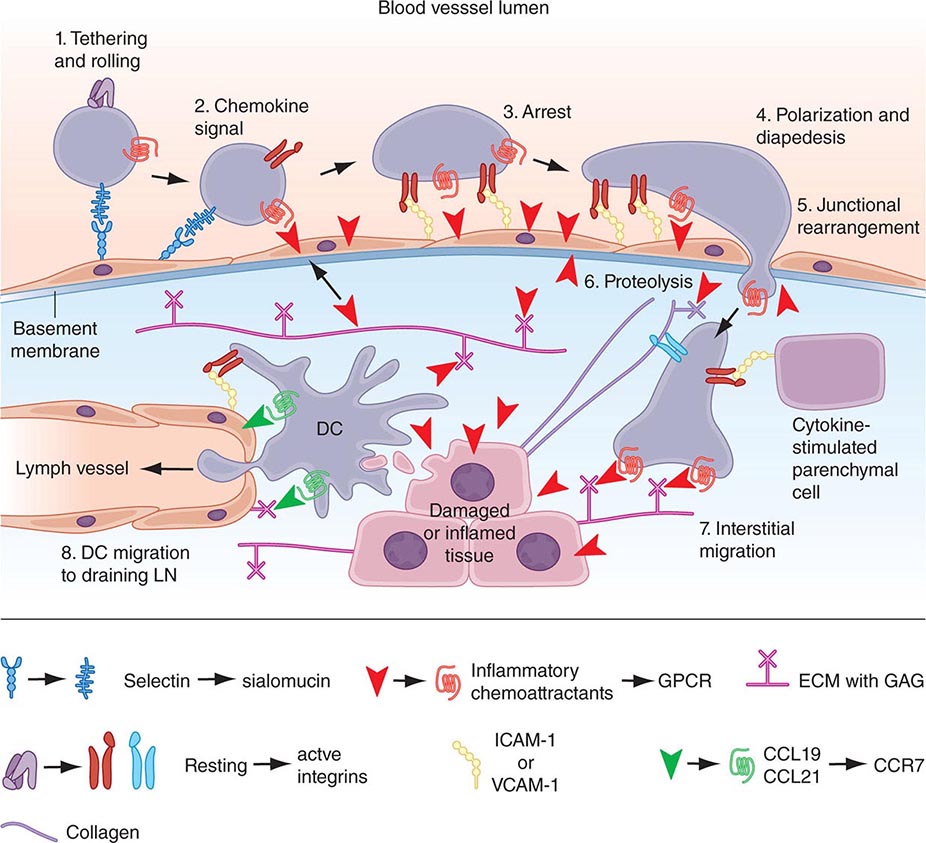
FIGURE 372e-11 Key migration steps of immune cells at sites of inflammation. Inflammation due to tissue damage or infection induces the release of cytokines (not shown) and inflammatory chemoattractants (red arrowheads) from distressed stromal cells and “professional” sentinels, such as mast cells and macrophages (not shown). The inflammatory signals induce upregulation of endothelial selectins and immunoglobulin “superfamily” members, particularly ICAM-1 and/or VCAM-1. Chemoattractants, particularly chemokines, are produced by or translocated across venular endothelial cells (red arrow) and are displayed in the lumen to rolling leukocytes. Those leukocytes that express the appropriate set of trafficking molecules undergo a multistep adhesion cascade (steps 1–3) and then polarize and move by diapedesis across the venular wall (steps 4 and 5). Diapedesis involves transient disassembly of endothelial junctions and penetration through the underlying basement membrane (step 6). Once in the extravascular (interstitial) space, the migrating cell uses different integrins to gain “footholds” on collagen fibers and other ECM molecules, such as laminin and fibronectin, and on inflammation-induced ICAM-1 on the surface of parenchymal cells (step 7). The migrating cell receives guidance cues from distinct sets of chemoattractants, particularly chemokines, which may be immobilized on glycosaminoglycans (GAG) that “decorate” many ECM molecules and stromal cells. Inflammatory signals also induce tissue dendritic cells (DCs) to undergo maturation. Once DCs process material from damaged tissues and invading pathogens, they upregulate CCR7, which allows them to enter draining lymph vessels that express the CCR7 ligand CCL21 (and CCL19). In lymph nodes (LNs), these antigen-loaded mature DCs activate naïve T cells and expand pools of effector lymphocytes, which enter the blood and migrate back to the site of inflammation. T cells in tissue also use this CCR7-dependent route to migrate from peripheral sites to draining lymph nodes through afferent lymphatics. (Adapted from AD Luster et al: Nat Immunol 6:1182, 2005; with permission from Macmillan Publishers Ltd. Copyright 2005.)
The first stage of lymphocyte–endothelial cell interactions, attachment and rolling, occurs when lymphocytes leave the stream of flowing blood cells in a postcapillary venule and roll along venule endothelial cells (Fig. 372e-11). Lymphocyte rolling is mediated by the L-selectin molecule (LECAM-1, LAM-1, CD62L) and slows cell transit time through venules, allowing time for activation of adherent cells.
The second stage of lymphocyte–endothelial cell interactions, firm adhesion with activation-dependent stable arrest, requires stimulation of lymphocytes by chemoattractants or by endothelial cell–derived cytokines. Cytokines thought to participate in adherent cell activation include members of the IL-8 family, platelet-activation factor, leukotriene B4, and C5a. In addition, HEVs express chemokines, SLC (CCL21) and ELC (CCL19), which participate in this process. Following activation by chemoattractants, lymphocytes shed L-selectin from the cell surface and upregulate cell CD11b/18 (MAC-1) or CD11a/18 (LFA-1) molecules, resulting in firm attachment of lymphocytes to HEVs.
Lymphocyte homing to peripheral lymph nodes involves adhesion of L-selectin to glycoprotein HEV ligands collectively referred to as peripheral node addressin (PNAd), whereas homing of lymphocytes to intestine Peyer’s patches primarily involves adhesion of the α4β7 integrin to mucosal addressin cell adhesion molecule-1 (MAdCAM-1) on the Peyer’s patch HEVs. However, for migration to mucosal Peyer’s patch lymphoid aggregates, naïve lymphocytes primarily use L-selectin, whereas memory lymphocytes use α4β7 integrin. α4β1 integrin (CD49d/CD29, VLA-4)–VCAM-1 interactions are important in the initial interaction of memory lymphocytes with HEVs of multiple organs in sites of inflammation (Table 372e-15).
TRAFFICKING MOLECULES INVOLVED IN INFLAMMATORY DISEASE PROCESSES |
The third stage of leukocyte emigration in HEVs is sticking and arrest. Sticking of the lymphocyte to endothelial cells and arrest at the site of sticking are mediated predominantly by ligation of α1β2 integrin LFA-1 to the integrin ligand ICAM-1 on HEVs. Whereas the first three stages of lymphocyte attachment to HEVs take only a few seconds, the fourth stage of lymphocyte emigration, transendothelial migration, takes ~10 min. Although the molecular mechanisms that control lymphocyte transendothelial migration are not fully characterized, the HEV CD44 molecule and molecules of the HEV glycocalyx (extracellular matrix) are thought to play important regulatory roles in this process (Fig. 372e-11). Finally, expression of matrix metalloproteases capable of digesting the subendothelial basement membrane, rich in nonfibrillar collagen, appears to be required for the penetration of lymphoid cells into the extravascular sites.
Abnormal induction of HEV formation and use of the molecules discussed above have been implicated in the induction and maintenance of inflammation in a number of chronic inflammatory diseases. In animal models of type 1 diabetes mellitus, MAdCAM-1 and GlyCAM-1 have been shown to be highly expressed on HEVs in inflamed pancreatic islets, and treatment of these animals with inhibitors of L-selectin and α4 integrin function blocked the development of type 1 diabetes mellitus (Chap. 417). A similar role for abnormal induction of the adhesion molecules of lymphocyte emigration has been suggested in rheumatoid arthritis (Chap. 380), Hashimoto’s thyroiditis (Chap. 405), Graves’ disease (Chap. 405), multiple sclerosis (Chap. 458), Crohn’s disease (Chap. 351), and ulcerative colitis (Chap. 351).
Immune-Complex Formation Clearance of antigen by immune-complex formation between antigen, complement, and antibody is a highly effective mechanism of host defense. However, depending on the level of immune complexes formed and their physicochemical properties, immune complexes may or may not result in host and foreign cell damage. After antigen exposure, certain types of soluble antigen-antibody complexes freely circulate and, if not cleared by the reticuloendothelial system, can be deposited in blood vessel walls and in other tissues such as renal glomeruli and cause vasculitis or glomerulonephritis syndromes (Chaps. 338 and 385). Deficiencies of early complement components are associated with inefficient clearance of immune complexes and immune complex mediated tissue damage in autoimmune syndromes, whereas deficiencies of the later complement components are associated with susceptibility to recurrent Neisseria infections (Table 372e-16).
COMPLEMENT DEFICIENCIES AND ASSOCIATED DISEASES |
Immediate-Type Hypersensitivity Helper T cells that drive antiallergen IgE responses are usually TH2-type inducer T cells that secrete IL-4, IL-5, IL-6, and IL-10. Mast cells and basophils have high-affinity receptors for the Fc portion of IgE (FcRI), and cell-bound antiallergen IgE effectively “arms” basophils and mast cells. Mediator release is triggered by antigen (allergen) interaction with Fc receptor-bound IgE, and the mediators released are responsible for the pathophysiologic changes of allergic diseases (Table 372e-11). Mediators released from mast cells and basophils can be divided into three broad functional types: (1) those that increase vascular permeability and contract smooth muscle (histamine, platelet-activating factor, SRS-A, BK-A), (2) those that are chemotactic for or activate other inflammatory cells (ECF-A, NCF, leukotriene B4), and (3) those that modulate the release of other mediators (BK-A, platelet-activating factor) (Chap. 376).
Cytotoxic Reactions of Antibody In this type of immunologic injury, complement-fixing (C1-binding) antibodies against normal or foreign cells or tissues (IgM, IgG1, IgG2, IgG3) bind complement via the classic pathway and initiate a sequence of events similar to that initiated by immune-complex deposition, resulting in cell lysis or tissue injury. Examples of antibody-mediated cytotoxic reactions include red cell lysis in transfusion reactions, Goodpasture’s syndrome with anti–glomerular basement membrane antibody formation, and pemphigus vulgaris with antiepidermal antibodies inducing blistering skin disease.
Classic Delayed-Type Hypersensitivity Reactions Inflammatory reactions initiated by mononuclear leukocytes and not by antibody alone have been termed delayed-type hypersensitivity reactions. The term delayed has been used to contrast a secondary cellular response that appears 48–72 h after antigen exposure with an immediate hypersensitivity response generally seen within 12 h of antigen challenge and initiated by basophil mediator release or preformed antibody. For example, in an individual previously infected with M. tuberculosis organisms, intradermal placement of tuberculin purified protein derivative as a skin test challenge results in an indurated area of skin at 48–72 h, indicating previous exposure to tuberculosis.
The cellular events that result in classic delayed-type hypersensitivity responses are centered on T cells (predominantly, although not exclusively, IFN-γ, IL-2, and TNF-α-secreting TH1-type helper T cells) and macrophages. Recently NK cells have been suggested to play a major role in the form of delayed hypersensitivity that occurs following skin contact with immunogens. First, local immune and inflammatory responses at the site of foreign antigen upregulate endothelial cell adhesion molecule expression, promoting the accumulation of lymphocytes at the tissue site. In the general schemes outlined in Figs. 372e-2 and 372e-3, antigen is processed by DCs and presented to small numbers of CD4+ T cells expressing a TCR specific for the antigen. IL-12 produced by APCs induces T cells to produce IFN-γ (TH1 response). Macrophages frequently undergo epithelioid cell transformation and fuse to form multinucleated giant cells in response to IFN-γ. This type of mononuclear cell infiltrate is termed granulomatous inflammation. Examples of diseases in which delayed-type hypersensitivity plays a major role are fungal infections (histoplasmosis; Chap. 236), mycobacterial infections (tuberculosis, leprosy; Chaps. 202 and 203), chlamydial infections (lymphogranuloma venereum; Chap. 213), helminth infections (schistosomiasis; Chap. 259), reactions to toxins (berylliosis; Chap. 311), and hypersensitivity reactions to organic dusts (hypersensitivity pneumonitis; Chap. 310). In addition, delayed-type hypersensitivity responses play important roles in tissue damage in autoimmune diseases such as rheumatoid arthritis, temporal arteritis, and granulomatosis with polyangiitis (Wegener’s) (Chaps. 380 and 385).
CLINICAL EVALUATION OF IMMUNE FUNCTION
Clinical assessment of immunity requires investigation of the four major components of the immune system that participate in host defense and in the pathogenesis of autoimmune diseases: (1) humoral immunity (B cells); (2) cell-mediated immunity (T cells, monocytes); (3) phagocytic cells of the reticuloendothelial system (macrophages), as well as polymorphonuclear leukocytes; and (4) complement. Clinical problems that require an evaluation of immunity include chronic infections, recurrent infections, unusual infecting agents, and certain autoimmune syndromes. The type of clinical syndrome under evaluation can provide information regarding possible immune defects (Chap. 374). Defects in cellular immunity generally result in viral, mycobacterial, and fungal infections. An extreme example of deficiency in cellular immunity is AIDS (Chap. 226). Antibody deficiencies result in recurrent bacterial infections, frequently with organisms such as S. pneumoniae and Haemophilus influenzae (Chap. 374). Disorders of phagocyte function are frequently manifested by recurrent skin infections, often due to Staphylococcus aureus (Chap. 80). Finally, deficiencies of early and late complement components are associated with autoimmune phenomena and recurrent Neisseria infections (Table 372e-16). For further discussion of useful initial screening tests of immune function, see Chap. 374.
IMMUNOTHERAPY
Many therapies for autoimmune and inflammatory diseases involve the use of nonspecific immune-modulating or immunosuppressive agents such as glucocorticoids or cytotoxic drugs. The goal of development of new treatments for immune-mediated diseases is to design ways to specifically interrupt pathologic immune responses, leaving nonpathologic immune responses intact. Novel ways to interrupt pathologic immune responses that are under investigation include the use of anti-inflammatory cytokines or specific cytokine inhibitors as anti-inflammatory agents, the use of monoclonal antibodies against T or B lymphocytes as therapeutic agents, the use of intravenous Ig for certain infections and immune complex–mediated diseases, the use of specific cytokines to reconstitute components of the immune system, and bone marrow transplantation to replace the pathogenic immune system with a more normal immune system (Chaps. 80, 374, and 226). In particular, the use of a monoclonal antibody to B cells (rituximab, anti-CD20 MAb) is approved in the United States for the treatment of non-Hodgkin’s lymphoma (Chap. 134) and, in combination with methotrexate, for treatment of adult patients with severe rheumatoid arthritis resistant to TNF-α inhibitors (Chap. 380). The U.S. Food and Drug Administration (FDA) approved the use of CTLA-4 antibodies in 2010 to block T cell anergy for use in cancer immunotherapy, and it was the first agent to demonstrate survival benefit in patients with advanced melanoma. Early-stage clinical trials have now shown that PD-1 blockade to reverse T cell exhaustion can induce tumor regression.
Cell-based therapies have been studied for many years, including ex vivo activation of NK cells for reinfusion into patients with malignancies and DC therapy of ex vivo priming of DCs for enhanced presentation of cancer antigens, with reinfusion of primed DCs into the patient. One such strategy for DC therapy has been approved by the FDA for treatment of advanced prostate cancer.
Cytokines and Cytokine Inhibitors Several TNF inhibitors are used as biological therapies in the treatment of rheumatoid arthritis; these include monoclonal antibodies, TNF-R Fc fusion proteins, and Fab fragments. Use of anti-TNF-α antibody therapies such as adalimumab, infliximab, and golimumab has resulted in clinical improvement in patients with these diseases and has opened the way for targeting TNF-α to treat other severe forms of autoimmune and/or inflammatory disease. Blockage of TNF-α has been effective in rheumatoid arthritis, psoriasis, Crohn’s disease, and ankylosing spondylitis. Other cytokine inhibitors are recombinant soluble TNF-α receptor (R) fused to human Ig and anakinra (soluble IL-1 receptor antagonist, or IL-1ra). The treatment of autoinflammatory syndromes (Table 372e-6) with recombinant IL-1 receptor antagonist can prevent symptoms in these syndromes, because the overproduction of IL-1β is a hallmark of these diseases.
TNF-αR-Fc fusion protein (etanercept) and IL-1ra act to inhibit the activity of pathogenic cytokines in rheumatoid arthritis, i.e., TNF-α and IL-1, respectively. Similarly, anti-IL-6, IFN-β, and IL-11 act to inhibit pathogenic proinflammatory cytokines. Anti-IL-6 (tocilizumab) inhibits IL-6 activity, whereas IFN-β and IL-11 decrease IL-1 and TNF-α production.
Of particular note has been the successful use of IFN-γ in the treatment of the phagocytic cell defect in chronic granulomatous disease (Chap. 80).
Monoclonal Antibodies to T and B Cells The OKT3 MAb against human T cells has been used for several years as a T cell-specific immunosuppressive agent that can substitute for horse antithymocyte globulin (ATG) in the treatment of solid organ transplant rejection. OKT3 produces fewer allergic reactions than ATG but does induce human anti-mouse Ig antibody—thus limiting its use. Anti-CD4 MAb therapy has been used in trials to treat patients with rheumatoid arthritis. While inducing profound immunosuppression, anti-CD4 MAb treatment also induces susceptibility to severe infections. Treatment of patients with a MAb against the T cell molecule CD40 ligand (CD154) is under investigation to induce tolerance to organ transplants, with promising results reported in animal studies. Monoclonal antibodies to the CD25 (IL-2a) receptor (basiliximab) are being used for treatment of graft-versus-host disease in bone marrow transplantation, and anti-CD20 MAb (rituximab) is used to treat hematologic neoplasms, autoimmune diseases, kidney transplant rejection, and rheumatoid arthritis. The anti-IgE monoclonal antibody (omalizumab) is used for blocking antigen-specific IgE that causes hay fever and allergic rhinitis (Chap. 376); however, side effects of anti-IgE include increased risk of anaphylaxis. Studies have shown that TH17 cells, in addition to TH1, are mediators of inflammation in Crohn’s disease, and anti–IL-12/IL-23p40 antibody therapy has been studied as a treatment.
It is important to realize the potential risks of these immunosuppressive monoclonal antibodies. Natalizumab is a humanized IgG antibody against an α4 integrin that inhibits leukocyte migration into tissues and has been approved for treatment of multiple sclerosis in the United States. Both it and anti-CD20 (rituximab) have been associated with the onset of progressive multifocal leukoencephalopathy (PML)—a serious and usually fatal CNS infection caused by JC polyomavirus. Efalizumab, a humanized IgG monoclonal antibody previously approved for treatment of plaque psoriasis, has now been taken off the market due to reactivation of JC virus leading to fatal PML. Thus, use of any currently approved immunosuppressant immunotherapies should be undertaken with caution and with careful monitoring of patients according to FDA guidelines.
Tolerance Induction Specific immunotherapy has moved into a new era with the introduction of soluble CTLA-4 protein into clinical trials. Use of this molecule to block T cell activation via TCR/CD28 ligation during organ or bone marrow transplantation has showed promising results in animals and in early human clinical trials. Specifically, treatment of bone marrow with CTLA-4 protein reduces rejection of the graft in HLA-mismatched bone marrow transplantation. In addition, promising results with soluble CTLA-4 have been reported in the downmodulation of autoimmune T cell responses in the treatment of psoriasis; and it is being studied for treatment of systemic lupus erythematosus (Chap. 378).
Intravenous Immunoglobulin (IVIg) IVIg has been used successfully to block reticuloendothelial cell function and immune complex clearance in various immune cytopenias such as immune thrombocytopenia (Chap. 140). In addition, IVIg is useful for prevention of tissue damage in certain inflammatory syndromes such as Kawasaki disease (Chap. 385) and as Ig replacement therapy for certain types of immunoglobulin deficiencies (Chap. 374). In addition, controlled clinical trials support the use of IVIg in selected patients with graft-versus-host disease, multiple sclerosis, myasthenia gravis, Guillain-Barré syndrome, and chronic demyelinating polyneuropathy.
Stem Cell Transplantation Hematopoietic stem cell transplantation (SCT) is now being comprehensively studied to treat several autoimmune diseases, including systemic lupus erythematosus, multiple sclerosis, and scleroderma. The goal of immune reconstitution in autoimmune disease syndromes is to replace a dysfunctional immune system with a normally reactive immune cell repertoire. Preliminary results in patients with scleroderma and lupus have showed encouraging results. Controlled clinical trials in these three diseases are now being launched in the United States and Europe to compare the toxicity and efficacy of conventional immunosuppression therapy with that of myeloablative autologous SCT. Recently, SCT was used in the setting of HIV-1 infection. HIV-1 infection of CD4+ T cells requires the presence of surface CD4 receptor and the chemokine receptor 5 (CCR5) co-receptor. Studies have demonstrated that patients who are homozygous for a 32-bp deletion in the CCR5 allele do not express CD4+ T cell CCR5 and thus are resistant to HIV-1 infection with HIV-1 strains that use this co-receptor. Stem cells from a homozygous CCR5 delta32 donor were transplanted to an HIV-infected patient following standard conditioning for such transplants, and the patient has maintained long-term control of the virus without antiretrovirals. Thus, a number of recent insights into immune system function have spawned a new field of interventional immunotherapy and have enhanced the prospect for development of more specific and nontoxic therapies for immune and inflammatory diseases.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


