Primary Cutaneous Anaplastic Large Cell Lymphoma
Aaron Auerbach, MD, PhD
Key Facts
Terminology
One of the primary cutaneous CD30(+) T-cell lymphoproliferative disorders
T-cell lymphoma of atypical, usually large CD30(+) cells, without evidence of mycosis fungoides
Clinical Issues
Solitary or localized nodules that can regress but frequently occur
Can involve regional lymph nodes
Treated with skin-targeted therapy for low-stage disease, chemotherapy for systemic disease
Good prognosis, ˜ 90% 10-year survival
Microscopic Pathology
Polymorphic background infiltrate not prominent
Diffuse sheets of large T cells in the dermis ± subcutis
Hallmark cells with multiple nuclei (horseshoe-shaped), helpful if present
Ancillary Tests
Immunohistochemistry: CD30(+), CD15(−), CD3(+), cytotoxic markers(+), EMA(−), ALK(−), CLA(+)
Molecular: No ALK gene translocations on chromosome 2
Top Differential Diagnoses
Systemic ALCL with cutaneous involvement
EMA(+), ALK(+) and ALK gene translocations, unlike primary cutaneous ALCL
Lymphomatoid papulosis (LyP)
Groups of papules that regress and recur
Wedge-shaped and prominent polymorphous background infiltrate
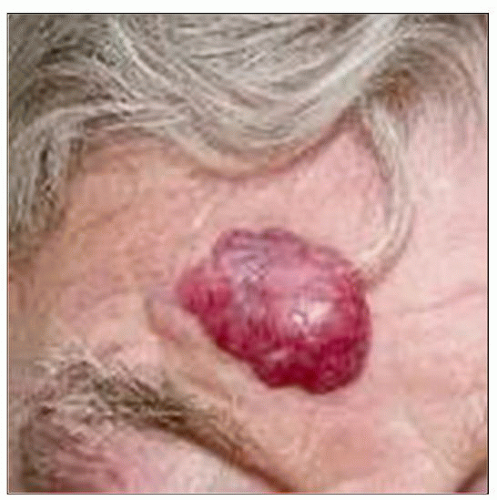 This is a solitary raised nonulcerated tumor nodule on the forehead of a man diagnosed with primary cutaneous anaplastic large cell lymphoma. (Courtesy R. Willemze, MD.) |
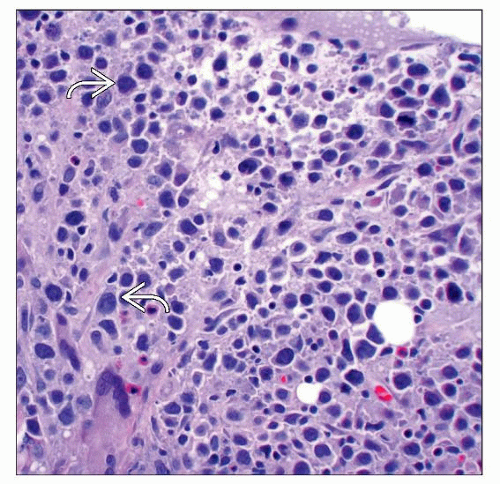 Histologic section of cutaneous ALCL shows large atypical cells with abundant cytoplasm and irregular multilobated nuclei  , admixed with a background of small inflammatory cells. , admixed with a background of small inflammatory cells. |
TERMINOLOGY
Abbreviations
Primary cutaneous anaplastic large cell lymphoma (PCALCL)
Synonyms
Regressing atypical histiocytosis
Primary cutaneous large cell T-cell lymphoma, CD30(+)
Definitions
CD30(+) T-cell lymphoma consisting of atypical, usually large cells, without evidence of mycosis fungoides
One of the primary cutaneous CD30(+) T-cell lymphoproliferative disorders, including lymphomatoid papulosis (LyP) and borderline lesions
ETIOLOGY/PATHOGENESIS
Idiopathic
Viral infection, chronic antigen stimulation, and immunosuppression may play a role
Mechanism may involve CD30/TRAF1 upregulation of NF-κB
CLINICAL ISSUES
Epidemiology
Incidence
2nd most common cutaneous T-cell lymphoma after mycosis fungoides
0.1-0.2 per 100,000 people
20% of systemic ALCL cases involve skin
Age
Median age: 60s, but can occur in children
Gender
Male:female = 2-3:1
Site
Often trunk, face, and extremities
Presentation
Solitary or localized nodules, tumors, or papules
± ulceration
Multifocal ˜ 20%
Usually no symptoms other than mass effect
Natural History
Partial or complete spontaneous regression (40%), but frequently recur
Extracutaneous dissemination ˜ 10%
Usually to regional lymph nodes
Treatment
Adjuvant therapy
Radiation for localized nodules
Methotrexate for multifocal lesions
Chemotherapy for systemic disease
Prognosis
Better than most T-cell lymphomas
˜ 90% 10-year survival
Age < 60 years and spontaneous regression are good prognostic indicators
Systemic disease is poor prognostic indicator
Multifocal skin lesions and local lymph node involvement do not yield worse prognosis
MICROSCOPIC PATHOLOGY
Histologic Features
Diffuse sheets of large T cells in the dermis ± subcutis
Infrequent epidermotropism ± ulceration
Can involve lymphatic spaces
Tumor cells
Large anaplastic, pleomorphic, or immunoblastic appearance
Anaplastic cells with roundish shapes ± abundant cytoplasm
Small cell and histiocyte-rich variants are rare
Mitotic figures
Hallmark cells with multiple nuclei (horseshoe-shaped), often not seen, but helpful if present
Polymorphic background infiltrate (eosinophils and plasma cells) uncommon, unlike LyP
Exceptions
Ulcerating ALCL has polymorphic infiltrate, fewer CD30(+) cells, and epidermal hyperplasia
Neutrophil-rich (pyogenic ALCL) shows clusters of neutrophils with only scattered CD30(+) cells
ANCILLARY TESTS
Immunohistochemistry
CD30(+) (> 75% of tumor cells), CD15(−)
T-cell antigens expressed (CD2[+], CD3[+], CD5[+], CD7[+])
But can show loss of T-cell antigens
CD7 most common
CD4(+)/CD8(−)
CD8(+) and CD4(−)/CD8(−) rarely
Cytotoxic markers positive
Granzyme-B, perforin, TIA
EMA(−) and ALK(−) in PCALCL
Usually EMA(+) and ALK(+) in systemic ALCL
ALK(+) skin tumor likely indicates secondary cutaneous ALCL
Cutaneous lymphocyte antigen (CLA[+]) and HOXC5(+) in PCALCL
But CLA(−) in systemic ALCL
Negative for B-cell markers (CD20 and CD79)
Rarely positive for pax-5
Cytogenetics
No specific findings
Molecular Genetics
Clonal T-cell receptor gene rearrangement ˜ 90%
No translocations of ALK gene on chromosome 2p23
DIFFERENTIAL DIAGNOSIS
Systemic ALCL with Cutaneous Involvement
Similar morphologic features and overlapping phenotype with PCALCL
Separate disease from PCALCL, with different molecular findings and prognosis
Primary tumor in extracutaneous site that secondarily involves skin
Differences from PCALCL
Systemic ALCL also found in extracutaneous sites
Typically EMA(+) and ALK(+), unlike PCALCL
Translocations involving ALK gene
Less favorable prognosis (especially ALK[-] systemic ALCL)
Lymphomatoid Papulosis (LyP)
Also CD30(+) T-cell lymphoproliferative disorder, but with more polymorphic background infiltrate
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


